
cif2indmf.py -w -q 14 -m 1_3_Sr2IrO4.mcif
------ ------------------------------------------------------------
0 average GF, non-correlated
1 |j,mj> basis, no symmetry, except time reversal (-jz=jz)
-1 |j,mj> basis, no symmetry, not even time reversal (-jz=jz)
2 real harmonics basis, no symmetry, except spin (up=dn)
-2 real harmonics basis, no symmetry, not even spin (up=dn)
3 t2g orbitals
-3 eg orbitals
4 |j,mj>, only l-1/2 and l+1/2
5 axial symmetry in real harmonics
6 hexagonal symmetry in real harmonics
7 cubic symmetry in real harmonics
8 axial symmetry in real harmonics, up different than down
9 hexagonal symmetry in real harmonics, up different than down
10 cubic symmetry in real harmonics, up different then down
11 |j,mj> basis, non-zero off diagonal elements
12 real harmonics, non-zero off diagonal elements
13 J_eff=1/2 basis for 5d ions, non-magnetic with symmetry
14 J_eff=1/2 basis for 5d ions, no symmetry
------ ------------------------------------------------------------"""
init_proj.py -a szero.py
In the first step we set-up wien2k calculation
init_lapw
Next we converge the DFT charge
run_lapw
initso
run_lapw -so
init_dmft.py
There are 56 atoms in the unit cell: 1 Ir1 2 Ir1 3 Ir1 4 Ir1 5 Ir2 6 Ir2 7 Ir2 8 Ir2 ... Specify correlated atoms (ex: 1-4,7,8): 1-8
For each atom, specify correlated orbital(s) (ex: d,f): 1 Ir1-1 d 2 Ir1-2 d 3 Ir1-3 d 4 Ir1-4 d 5 Ir2-5 d 6 Ir2-6 d 7 Ir2-7 d 8 Ir2-8 d
Specify qsplit for each correlated orbital (default = 0):
Qsplit Description
------ ------------------------------------------------------------
0 average GF, non-correlated
1 |j,mj> basis, no symmetry, except time reversal (-jz=jz)
-1 |j,mj> basis, no symmetry, not even time reversal (-jz=jz)
2 real harmonics basis, no symmetry, except spin (up=dn)
-2 real harmonics basis, no symmetry, not even spin (up=dn)
3 t2g orbitals
-3 eg orbitals
4 |j,mj>, only l-1/2 and l+1/2
.....
------ ------------------------------------------------------------
1 Ir1-1 d: 3
2 Ir1-2 d: 3
3 Ir1-3 d: 3
4 Ir1-4 d: 3
5 Ir2-5 d: 3
6 Ir2-6 d: 3
7 Ir2-7 d: 3
8 Ir2-8 d: 3
Specify projector type (default = 2):
Projector Description
------ ------------------------------------------------------------
1 projection to the solution of Dirac equation (to the head)
2 projection to the Dirac solution, its energy derivative,
LO orbital, as described by P2 in PRB 81, 195107 (2010)
4 similar to projector-2, but takes fixed number of bands in
some energy range, even when chemical potential and
MT-zero moves (folows band with certain index)
5 fixed projector, which is written to projectorw.dat. You can
generate projectorw.dat with the tool wavef.py
------ ------------------------------------------------------------
> 5
Do you want to group any of these orbitals into cluster-DMFT problems? (y/n): n
Enter the correlated problems forming each unique correlated problem, separated by spaces (ex: 1,3 2,4 5-8): 1-8
Range (in eV) of hybridization taken into account in impurity problems; default -10.0, 10.0: < ENTER >
Perform calculation on real; or imaginary axis? (r/i): r
Is this a spin-orbit run? (y/n): y
The script init_dmft.py generates Sr2IrO4.indmfl, Sr2IrO4.indmfi and projectorw.dat. Let us take a closer look at the header of the Sr2IrO4.indmfl file:
369 832 1 5 # hybridization band index nemin and nemax, renormalize for interstitials, projection type 0 0.025 0.025 200 -3.000000 1.000000 # matsubara, broadening-corr, broadening-noncorr, nomega, omega_min, omega_max (in eV) 8 # number of correlated atoms 1 1 0 # iatom, nL, locrot 2 3 1 # L, qsplit, cix 2 1 0 # iatom, nL, locrot 2 3 2 # L, qsplit, cix 3 1 0 # iatom, nL, locrot 2 3 3 # L, qsplit, cix 4 1 0 # iatom, nL, locrot 2 3 4 # L, qsplit, cix 5 1 0 # iatom, nL, locrot 2 3 5 # L, qsplit, cix 6 1 0 # iatom, nL, locrot 2 3 6 # L, qsplit, cix 7 1 0 # iatom, nL, locrot 2 3 7 # L, qsplit, cix 8 1 0 # iatom, nL, locrot 2 3 8 # L, qsplit, cix
The second line shows that we are performing calculation on real axis (at the moment) and also gives k-point broadening.
Next several lines contain declaration for all 8 correlated atoms. There are two lines per atom, and the first line contains atom index, number of L-types, and locrot. This last integer (locrot) will be now changed to apply local rotations to Ir atoms, such that the local axis will coincide with octahedron cage.
The global axis of the structure is not convenient for DMFT
calculation. While in principle the DMFT is rotationally invariant
method, and any axis should give the same result, in practice there is
enormous difference in the severity of the quamtum Monte Carlo sign
problem. To minimize the sign problem, we always pick the local axis on
each atom so that the hybridization matrix is maximally diagonal. This
is usually the local axis, which is aligned with the local environment of
the correlated atom, in this case octahedron. In this figure, we show
how the local axis on Ir will be choosen below.
localaxes.py
1 ATOM: 1 EQUIV. 1 Ir1 AT 0.25000 0.00000 0.87500 2 ATOM: 1 EQUIV. 2 Ir1 AT 0.75000 0.00000 0.12500 3 ATOM: 1 EQUIV. 3 Ir1 AT 0.75000 0.00000 0.62500 4 ATOM: 1 EQUIV. 4 Ir1 AT 0.25000 0.00000 0.37500 5 ATOM: 2 EQUIV. 1 Ir2 AT 0.75000 0.50000 0.37500 6 ATOM: 2 EQUIV. 2 Ir2 AT 0.25000 0.50000 0.62500 7 ATOM: 2 EQUIV. 3 Ir2 AT 0.25000 0.50000 0.12500 8 ATOM: 2 EQUIV. 4 Ir2 AT 0.75000 0.50000 0.87500 ..... Enter the atom for which you want to determine local axes: 1
The script will find that we have octahedra, and it will calculate the local axis:
trying cube :-N= -2.0823405461 chi2= 0.120703936262 = 3.79142326447 combined-criteria 6.71874235103 trying octahedra : -N= -0.0823405495682 chi2= 1.48029736617e-17 = 3.79142326429 combined-criteria 0.0067799661032 trying tetrahedra : -N= 0.0 chi2= 0.582079772951 = 3.74769905636 combined-criteria 11.4897941778 trying square-piramid : -N= -0.0472857590573 chi2= 0.493480215373 = 3.77302040758 combined-criteria 9.743144954 According to angle variance and bond-angle we are trying octahedra with = 0.0823405495682 and = 1.48029736617e-17 Found the environment is ('octahedra', 0.082340549568209909, 1.4802973661668754e-17) Rotation to input into case.indmfl by locrot=-1 : -0.83256188 0.55393206 0.00000000 -0.55393206 -0.83256188 0.00000000 0.00000000 0.00000000 1.00000000
The set of atoms, which are equivalent in case.struct file, should use the same transformation to local axis. The reason is that the code will automatically find extra symmetry operation transforming first atom to any equivalent atom, and will add this extra transformation.
In our example of Sr2IrO4, we have two inequivalent atoms in structure file, and the second group of atoms starts with atom number 5. We hence need rotation for the atom nubmber 5 as well. To get it, we rerun the script
localaxes_new.py
Rotation to input into case.indmfl by locrot=-1 : 0.83256188 -0.55393206 -0.00000000 -0.55393206 -0.83256188 -0.00000000 -0.00000000 -0.00000000 -1.00000000
Now that we have the transformation matrix to local coordinate system, we will add it to Sr2IrO4.indmfl file. We will change locrot to -1 for the relevant atom, and paste below the transformation for that atom. The header of the Sr2IrO4.struct file will look like that
369 832 1 5 # hybridization band index nemin and nemax, renormalize for interstitials, projection type 0 0.025 0.025 200 -3.000000 1.000000 # matsubara, broadening-corr, broadening-noncorr, nomega, omega_min, omega_max (in eV) 8 # number of correlated atoms 1 1 -1 # iatom, nL, locrot 2 14 1 # L, qsplit, cix -0.83256188 0.55393206 0.00000000 -0.55393206 -0.83256188 0.00000000 0.00000000 0.00000000 1.00000000 2 1 -1 # iatom, nL, locrot 2 14 2 # L, qsplit, cix -0.83256188 0.55393206 0.00000000 -0.55393206 -0.83256188 0.00000000 0.00000000 0.00000000 1.00000000 3 1 -1 # iatom, nL, locrot 2 14 3 # L, qsplit, cix -0.83256188 0.55393206 0.00000000 -0.55393206 -0.83256188 0.00000000 0.00000000 0.00000000 1.00000000 4 1 -1 # iatom, nL, locrot 2 14 4 # L, qsplit, cix -0.83256188 0.55393206 0.00000000 -0.55393206 -0.83256188 0.00000000 0.00000000 0.00000000 1.00000000 5 1 -1 # iatom, nL, locrot 2 14 5 # L, qsplit, cix 0.83256188 -0.55393206 -0.00000000 -0.55393206 -0.83256188 -0.00000000 -0.00000000 -0.00000000 -1.00000000 6 1 -1 # iatom, nL, locrot 2 14 6 # L, qsplit, cix 0.83256188 -0.55393206 -0.00000000 -0.55393206 -0.83256188 -0.00000000 -0.00000000 -0.00000000 -1.00000000 7 1 -1 # iatom, nL, locrot 2 14 7 # L, qsplit, cix 0.83256188 -0.55393206 -0.00000000 -0.55393206 -0.83256188 -0.00000000 -0.00000000 -0.00000000 -1.00000000 8 1 -1 # iatom, nL, locrot 2 14 8 # L, qsplit, cix 0.83256188 -0.55393206 -0.00000000 -0.55393206 -0.83256188 -0.00000000 -0.00000000 -0.00000000 -1.00000000
We already removed the eg states from correlated subset, as the Sigind and Transformation matrix take the form:
#================ # Siginds and crystal-field transformations for correlated orbitals ================ 8 10 3 # Number of independent kcix blocks, max dimension, max num-independent-components 1 10 3 # cix-num, dimension, num-independent-components #---------------- # Independent components are -------------- 'xz' 'yz' 'xy' 'xz' 'yz' 'xy' #---------------- # Sigind follows -------------------------- 1 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 #---------------- # Transformation matrix follows ----------- 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 -0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.00000000 -0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 -0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.00000000 -0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000
We first create a blank self-energy on the real axis. As we do not have params.dat file yet, we will need to give more arguments to the script:
szero.py -N 200 -L 25 -x 0.05
x lapw0 -f Sr2Iro4 x_dmft.py lapw1 x_dmft.py lapwso
x_dmft.py dmft1
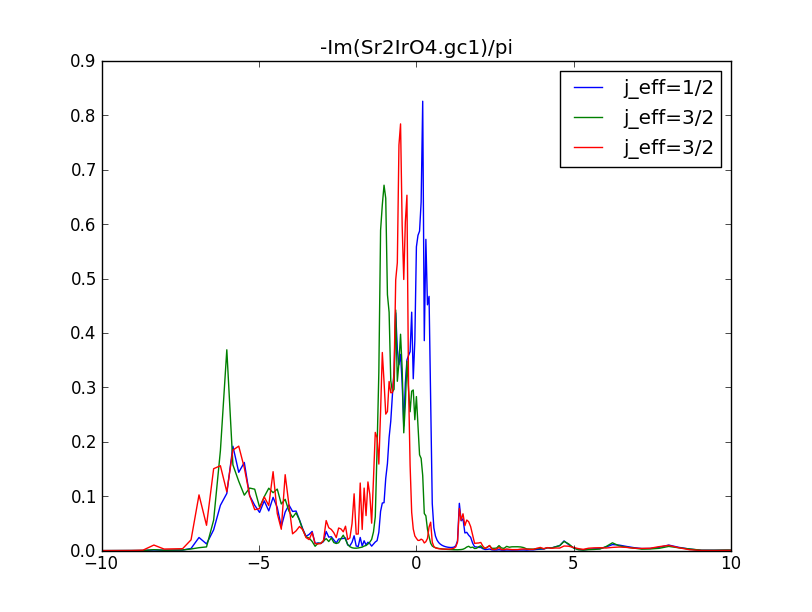
The spectral function, which is proportional to the imaginary part of
the green's function from Sr2IrO4.gc1, shows doubly
degenerate "xz" and "yz" states as well as the "xy" state near the
Fermi level:

findRot.py
#================ # Siginds and crystal-field transformations for correlated orbitals ================ 8 10 6 # Number of independent kcix blocks, max dimension, max num-independent-components 1 10 6 # cix-num, dimension, num-independent-components #---------------- # Independent components are -------------- 'xz' 'yz' 'xy' 'xz' 'yz' 'xy' #---------------- # Sigind follows -------------------------- 1 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 4 0 0 0 0 0 0 0 0 0 0 5 0 0 0 0 0 0 0 0 0 0 6 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 #---------------- # Transformation matrix follows ----------- -0.00000000 -0.00000000 -0.00005873 -0.00002664 -0.00000000 -0.00000000 0.83142944 -0.00000000 -0.00000000 -0.00000000 -0.39218728 -0.02348818 -0.00000000 -0.00000000 -0.00000000 -0.00000000 -0.00000000 -0.00000000 0.39218728 0.02348818 0.39218727 -0.02348816 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.39218727 0.02348816 0.00000000 0.00000000 0.83142944 0.00000000 0.00000000 0.00000000 -0.00005879 0.00002672 -0.00000000 -0.00000000 0.00000000 0.00000000 0.99999999 0.00000000 0.00000000 0.00000000 -0.00004935 0.00000270 0.00000000 0.00000000 -0.00012894 0.00002910 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00012894 -0.00002910 0.00012888 0.00002912 0.00000000 -0.00000000 -0.00000000 -0.00000000 0.00000000 -0.00000000 -0.00012888 -0.00002912 0.00000000 -0.00000000 -0.00004924 -0.00000261 -0.00000000 -0.00000000 0.99999999 -0.00000000 -0.00000000 0.00000000 0.58790938 -0.00000000 0.00000000 0.00000000 -0.00000000 0.00000000 0.00000000 0.00000000 -0.58790938 -0.00000000 0.00000000 0.00000000 -0.55463656 -0.03321728 -0.00000000 0.00000000 -0.00017894 0.00003406 -0.00000000 -0.00000000 -0.00000000 0.00000000 0.00017907 0.00003407 -0.00000000 0.00000000 0.55463657 -0.03321730 -0.00000000 0.00000000 0.58790938 -0.00000000 -0.00000000 0.00000000 -0.00000000 0.00000000 -0.00000000 0.00000000 -0.58790938 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000
Since this script is rather new, we also explain how to obtain the same transformation in a series of steps, which the script performs.
icix= 1 at omega=0
-0.37570022 0.00000000 0.00001700 -0.18308321 -0.00000000 -0.00000000 -0.00000000 0.00000000 0.00000000 0.00000000 -0.01462619 -0.24425465
0.00001700 0.18308321 -0.37578835 -0.00000000 -0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.00000000 0.24425831 -0.01463095
-0.00000000 0.00000000 -0.00000000 -0.00000000 -0.47922081 -0.00000000 0.01462616 0.24425466 -0.24425827 0.01463096 0.00000000 0.00000000
-0.00000000 -0.00000000 0.00000000 -0.00000000 0.01462616 -0.24425466 -0.37570020 -0.00000000 0.00001704 0.18308320 -0.00000000 -0.00000000
-0.00000000 -0.00000000 0.00000000 0.00000000 -0.24425827 -0.01463096 0.00001704 -0.18308320 -0.37578836 0.00000000 0.00000000 0.00000000
-0.01462619 0.24425465 0.24425831 0.01463095 0.00000000 -0.00000000 -0.00000000 0.00000000 0.00000000 -0.00000000 -0.47922081 -0.00000000
strHc="""
-0.37536788 0.00000000 0.00000523 -0.18586706 0.00003407 0.00000168 0.00000000 0.00000000 -0.00001470 -0.00001043 -0.37735904 -0.02945573
0.00000523 0.18586706 -0.37537965 -0.00000000 -0.00002855 -0.00000926 0.00001470 0.00001043 0.00000000 0.00000000 0.02945245 -0.37735839
0.00003407 -0.00000168 -0.00002855 0.00000926 -0.73087107 -0.00000000 -0.37735902 -0.02945575 0.02945245 -0.37735838 -0.00000000 -0.00000000
0.00000000 -0.00000000 0.00001470 -0.00001043 -0.37735902 0.02945575 -0.37536738 -0.00000000 0.00000524 0.18586689 -0.00003407 0.00000168
-0.00001470 0.00001043 0.00000000 -0.00000000 0.02945245 0.37735838 0.00000524 -0.18586689 -0.37537917 0.00000000 0.00002855 -0.00000926
-0.37735904 0.02945573 0.02945245 0.37735839 -0.00000000 0.00000000 -0.00003407 -0.00000168 0.00002855 0.00000926 -0.73087055 0.00000000
"""
strT2C="""
0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 -0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000
0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000
-0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000
0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 -0.70710679 0.00000000 0.00000000 0.00000000
0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000
0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000
0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000
0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000
0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000
0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000
"""
We then execute the python script find5dRotation.py
find5dRotation.py diag_params.dat
Rotation to input : -0.00000000 -0.00000000 -0.00005873 -0.00002664 -0.00000000 -0.00000000 0.83142944 -0.00000000 -0.00000000 -0.00000000 -0.39218728 -0.02348818 -0.00000000 -0.00000000 -0.00000000 -0.00000000 -0.00000000 -0.00000000 0.39218728 0.02348818 0.39218727 -0.02348816 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.39218727 0.02348816 0.00000000 0.00000000 0.83142944 0.00000000 0.00000000 0.00000000 -0.00005879 0.00002672 -0.00000000 -0.00000000 0.00000000 0.00000000 0.99999999 0.00000000 0.00000000 0.00000000 -0.00004935 0.00000270 0.00000000 0.00000000 -0.00012894 0.00002910 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00012894 -0.00002910 0.00012888 0.00002912 0.00000000 -0.00000000 -0.00000000 -0.00000000 0.00000000 -0.00000000 -0.00012888 -0.00002912 0.00000000 -0.00000000 -0.00004924 -0.00000261 -0.00000000 -0.00000000 0.99999999 -0.00000000 -0.00000000 0.00000000 0.58790938 -0.00000000 0.00000000 0.00000000 -0.00000000 0.00000000 0.00000000 0.00000000 -0.58790938 -0.00000000 0.00000000 0.00000000 -0.55463656 -0.03321728 -0.00000000 0.00000000 -0.00017894 0.00003406 -0.00000000 -0.00000000 -0.00000000 0.00000000 0.00017907 0.00003407 -0.00000000 0.00000000 0.55463657 -0.03321730 -0.00000000 0.00000000 0.58790938 -0.00000000 -0.00000000 0.00000000 -0.00000000 0.00000000 -0.00000000 0.00000000 -0.58790938 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000
This transformation should now replace the transformation matrix in Sr2IrO4.indmfl file. The latter should now have the following form
#================ # Siginds and crystal-field transformations for correlated orbitals ================ 8 10 3 # Number of independent kcix blocks, max dimension, max num-independent-components 1 10 3 # cix-num, dimension, num-independent-components #---------------- # Independent components are -------------- 'xz' 'yz' 'xy' 'xz' 'yz' 'xy' #---------------- # Sigind follows -------------------------- 1 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 #---------------- # Transformation matrix follows ----------- -0.00000000 -0.00000000 -0.00005873 -0.00002664 -0.00000000 -0.00000000 0.83142944 -0.00000000 -0.00000000 -0.00000000 -0.39218728 -0.02348818 -0.00000000 -0.00000000 -0.00000000 -0.00000000 -0.00000000 -0.00000000 0.39218728 0.02348818 0.39218727 -0.02348816 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.39218727 0.02348816 0.00000000 0.00000000 0.83142944 0.00000000 0.00000000 0.00000000 -0.00005879 0.00002672 -0.00000000 -0.00000000 0.00000000 0.00000000 0.99999999 0.00000000 0.00000000 0.00000000 -0.00004935 0.00000270 0.00000000 0.00000000 -0.00012894 0.00002910 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00012894 -0.00002910 0.00012888 0.00002912 0.00000000 -0.00000000 -0.00000000 -0.00000000 0.00000000 -0.00000000 -0.00012888 -0.00002912 0.00000000 -0.00000000 -0.00004924 -0.00000261 -0.00000000 -0.00000000 0.99999999 -0.00000000 -0.00000000 0.00000000 0.58790938 -0.00000000 0.00000000 0.00000000 -0.00000000 0.00000000 0.00000000 0.00000000 -0.58790938 -0.00000000 0.00000000 0.00000000 -0.55463656 -0.03321728 -0.00000000 0.00000000 -0.00017894 0.00003406 -0.00000000 -0.00000000 -0.00000000 0.00000000 0.00017907 0.00003407 -0.00000000 0.00000000 0.55463657 -0.03321730 -0.00000000 0.00000000 0.58790938 -0.00000000 -0.00000000 0.00000000 -0.00000000 0.00000000 -0.00000000 0.00000000 -0.58790938 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 ...... ......
Please note that the orbitals are now doubly degenerate, and
consequently there are only 3 inequivalent orbitals. The paramter
"max num-independent-components"
and "num-independent-components" was consequently changed from
6 to 3.
szero.py
If vector file still exists, we might need to execute only the command
x_dmft.py dmft1
icix= 1 at omega=0
0.03859820 -0.00000000 0.00000000 -0.00000000 0.00000000 0.00000001 -0.00000000 -0.00000000 -0.00000000 0.00000000 -0.00000000 -0.00000000
0.00000000 0.00000000 0.03859818 0.00000000 0.00000000 -0.00000000 -0.00000000 -0.00000000 0.00000000 -0.00000000 0.00000000 -0.00000000
0.00000000 -0.00000001 0.00000000 0.00000000 -0.55882749 -0.00000000 0.00000000 0.00000000 0.00000000 -0.00000000 0.00000000 -0.00000000
-0.00000000 0.00000000 -0.00000000 0.00000000 0.00000000 -0.00000000 -0.55882748 -0.00000000 -0.00000000 -0.00000000 0.00000000 0.00000000
-0.00000000 -0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.00000000 0.00000000 -0.71048007 -0.00000000 -0.00000000 0.00000000
-0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.00000000 -0.00000000 -0.00000000 -0.71048008 -0.00000000
#---------------- # Sigind follows -------------------------- 1 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 4 0 0 0 0 0 0 0 0 0 0 5 0 0 0 0 0 0 0 0 0 0 6 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
#---------------- # Sigind follows -------------------------- 1 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
1 # number of sigind blocks 10 # dimension of this sigind block 1 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
Density matix= n1 0 0 0 0 0 0 0 n1 0 0 0 0 0 0 0 n2 0 0 0 0 0 0 0 n2 0 0 0 0 0 0 0 n3 0 0 0 0 0 0 0 n4 0 0 0 0 0 0 0 n4
local2global.py
Do you want to point magnetic moments in a new specified direction? [y/n]:n Do you want short or long output [s/l]? s Resuts written to output file moments.dat .
**** Correlated atom 1 ***** Mx_global= 0.00002456 -0.00000000 -0.69844846 -0.57517063 0.00000316 -0.00000069 1.03237132 -0.65279371 -0.61663046 -0.45228908 0.00006982 -0.00005030 0.86837984 -0.57821706 0.50136566 0.33384327 0.00007464 -0.00006166 0.00003205 -0.00003646 -0.69844846 0.57517063 -0.00002456 0.00000000 1.03237130 0.65279368 -0.00000316 -0.00000069 0.00006982 0.00005030 0.61663017 -0.45228888 -0.00007464 -0.00006166 -0.00003205 -0.00003646 0.86837999 0.57821716 0.50136576 -0.33384335 0.00000316 0.00000069 1.03237130 -0.65279368 0.00010713 -0.00000000 0.00002724 0.00000694 0.08928409 -0.01048992 0.00001064 -0.00000573 1.01944112 0.67879373 0.58856989 -0.39189466 0.00004411 0.00007345 -0.00002547 -0.00004240 1.03237132 0.65279371 -0.00000316 0.00000069 0.00002724 -0.00000694 -0.00010713 -0.00000000 0.00001064 0.00000573 -0.08928378 -0.01048972 -0.00004411 0.00007345 0.00002547 -0.00004240 1.01944113 -0.67879377 0.58856988 0.39189465 -0.61663046 0.45228908 0.00006982 -0.00005030 0.08928409 0.01048992 0.00001064 -0.00000573 0.00003726 0.00000000 0.05313166 -0.09436968 0.00002913 0.00007843 0.00003115 -0.00000157 -0.50470155 -0.39602562 -0.32333616 0.18066224 0.00006982 0.00005030 0.61663017 0.45228888 0.00001064 0.00000573 -0.08928378 0.01048972 0.05313166 0.09436968 -0.00003726 -0.00000000 0.50470182 -0.39602584 0.32333630 0.18066234 0.00002913 -0.00007843 0.00003115 0.00000157 0.86837984 0.57821706 -0.00007464 0.00006166 1.01944112 -0.67879373 -0.00004411 -0.00007345 0.00002913 -0.00007843 0.50470182 0.39602584 -0.00001058 -0.00000000 0.00000000 -0.00000000 0.83236365 -0.55422988 0.00000000 0.00000000 0.50136566 -0.33384327 -0.00003205 0.00003646 0.58856989 0.39189466 0.00002547 0.00004240 0.00003115 0.00000157 0.32333630 -0.18066234 0.00000000 -0.00000000 -0.00001058 0.00000000 -0.00000000 -0.00000000 0.83236367 -0.55422989 0.00007464 0.00006166 0.86837999 -0.57821716 0.00004411 -0.00007345 1.01944113 0.67879377 -0.50470155 0.39602562 0.00002913 0.00007843 0.83236365 0.55422988 -0.00000000 -0.00000000 0.00001058 -0.00000000 0.00000000 0.00000000 0.00003205 0.00003646 0.50136576 0.33384335 -0.00002547 0.00004240 0.58856988 -0.39189465 -0.32333616 -0.18066224 0.00003115 -0.00000157 0.00000000 -0.00000000 0.83236367 0.55422989 -0.00000000 -0.00000000 0.00001058 0.00000000 My_global= 0.00004264 -0.00000000 0.57522388 -0.69843801 0.00000424 0.00015729 -0.65276669 -1.03237702 0.45230126 -0.61662159 -0.00011231 -0.00007644 -0.57822314 -0.86840633 -0.33382780 0.50136832 -0.00004970 0.00004105 -0.00007786 0.00001986 0.57522388 0.69843801 -0.00004264 -0.00000000 -0.65276668 1.03237699 -0.00000424 0.00015729 -0.00011231 0.00007644 -0.45230107 -0.61662136 0.00004970 0.00004105 0.00007786 0.00001986 -0.57822326 0.86840653 -0.33382785 -0.50136842 0.00000424 -0.00015729 -0.65276668 -1.03237699 0.00001265 0.00000000 0.00000695 -0.00002723 -0.01047193 -0.08928636 -0.00000106 -0.00000029 -0.67878667 1.01942512 -0.39190476 -0.58857009 0.00006624 0.00011030 -0.00003825 -0.00006368 -0.65276669 1.03237702 -0.00000424 -0.00015729 0.00000695 0.00002723 -0.00001265 0.00000000 -0.00000106 0.00000029 0.01047170 -0.08928603 -0.00006624 0.00011030 0.00003825 -0.00006368 -0.67878664 -1.01942511 -0.39190478 0.58857011 0.45230126 0.61662159 -0.00011231 -0.00007644 -0.01047193 0.08928636 -0.00000106 -0.00000029 0.00000319 0.00000000 -0.09437060 -0.05312998 -0.00001247 0.00003574 0.00006745 -0.00004974 0.39602833 -0.50468873 0.18066847 0.32333186 -0.00011231 0.00007644 -0.45230107 0.61662136 -0.00000106 0.00000029 0.01047170 0.08928603 -0.09437060 0.05312998 -0.00000319 0.00000000 -0.39602848 -0.50468901 -0.18066858 0.32333204 -0.00001247 -0.00003574 0.00006745 0.00004974 -0.57822314 0.86840633 0.00004970 -0.00004105 -0.67878667 -1.01942512 -0.00006624 -0.00011030 -0.00001247 -0.00003574 -0.39602848 0.50468901 -0.00004707 0.00000000 0.00000000 -0.00000000 -0.55422987 -0.83236365 0.00000000 -0.00000000 -0.33382780 -0.50136832 0.00007786 -0.00001986 -0.39190476 0.58857009 0.00003825 0.00006368 0.00006745 0.00004974 -0.18066858 -0.32333204 0.00000000 0.00000000 -0.00004707 -0.00000000 0.00000000 -0.00000000 -0.55422989 -0.83236367 -0.00004970 -0.00004105 -0.57822326 -0.86840653 0.00006624 -0.00011030 -0.67878664 1.01942511 0.39602833 0.50468873 -0.00001247 0.00003574 -0.55422987 0.83236365 0.00000000 0.00000000 0.00004707 0.00000000 -0.00000000 -0.00000000 -0.00007786 -0.00001986 -0.33382785 0.50136842 -0.00003825 0.00006368 -0.39190478 -0.58857011 0.18066847 -0.32333186 0.00006745 -0.00004974 0.00000000 0.00000000 -0.55422989 0.83236367 0.00000000 0.00000000 0.00004707 0.00000000 Mz_global= -1.17689005 0.00000000 0.00006524 -0.00006784 -0.00000217 0.00000732 0.00012145 0.00000272 -0.00005380 0.00010349 -1.33452976 -0.10416443 0.00001803 0.00004699 0.00008675 -0.00002589 0.00000000 -0.00000000 -1.04443013 -0.08152121 0.00006524 0.00006784 1.17689093 0.00000000 0.00012145 -0.00000272 0.00000214 0.00000737 -1.33452933 0.10416445 0.00005380 0.00010349 -0.00000000 -0.00000000 1.04442956 -0.08152121 0.00001803 -0.00004699 0.00008675 0.00002589 -0.00000217 -0.00000732 0.00012145 0.00000272 0.00000001 0.00000000 0.00000000 0.00000000 -0.00003862 0.00004562 -0.00001023 -0.00001190 0.00002116 -0.00005517 -0.00002461 0.00002489 -0.00000000 -0.00000001 -0.00001575 -0.00001244 0.00012145 -0.00000272 0.00000214 -0.00000737 0.00000000 -0.00000000 -0.00000001 0.00000000 -0.00001023 0.00001193 0.00003862 0.00004562 0.00000000 -0.00000001 0.00001578 -0.00001244 0.00002116 0.00005517 -0.00002461 -0.00002489 -0.00005380 -0.00010349 -1.33452933 -0.10416445 -0.00003862 -0.00004562 -0.00001023 -0.00001193 -0.17689092 0.00000000 0.00004316 -0.00003740 0.00000000 -0.00000000 1.70367877 0.00000000 -0.00001330 0.00002795 -0.00000508 -0.00001713 -1.33452976 0.10416443 0.00005380 -0.00010349 -0.00001023 0.00001190 0.00003862 -0.00004562 0.00004316 0.00003740 0.17689007 0.00000000 0.00001330 0.00002795 0.00000508 -0.00001713 0.00000000 0.00000000 1.70367843 -0.00000000 0.00001803 -0.00004699 -0.00000000 0.00000000 0.00002116 0.00005517 0.00000000 0.00000001 0.00000000 0.00000000 0.00001330 -0.00002795 -1.00000000 0.00000000 -0.00000000 0.00000000 0.00001728 0.00004504 0.00000000 0.00000000 0.00008675 0.00002589 1.04442956 0.08152121 -0.00002461 -0.00002489 0.00001578 0.00001244 1.70367877 -0.00000000 0.00000508 0.00001713 0.00000000 0.00000000 -1.00000002 0.00000000 -0.00000000 0.00000000 0.00001728 0.00004504 0.00000000 0.00000000 0.00001803 0.00004699 -0.00000000 0.00000001 0.00002116 -0.00005517 -0.00001330 -0.00002795 0.00000000 -0.00000000 0.00001728 -0.00004504 0.00000000 -0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 -1.04443013 0.08152121 0.00008675 -0.00002589 -0.00001575 0.00001244 -0.00002461 0.00002489 -0.00000508 0.00001713 1.70367843 0.00000000 0.00000000 -0.00000000 0.00001728 -0.00004504 0.00000000 -0.00000000 1.00000002 0.00000000
atom1: [ 1, 1,0] atom2: [-1,-1,0] atom3: [-1,-1,0] atom4: [ 1, 1,0] atom5: [ 1,-1,0] atom6: [-1, 1,0] atom7: [-1, 1,0] atom8: [ 1,-1,0]
Now we just need to rerun local2global.py and provide the script with that information:
local2global.py
Do you want to point magnetic moments in a new specified direction? [y/n]:y
*** Please give your answer in python notaions, such as the example: [1,1,1]
Valid answer is also None, which does not change the current direction
Direction for atom Ir1[ 0] 0.2500 0.0000 0.8750 ? [1,1,0]
Direction for atom Ir1[ 1] 0.7500 0.0000 0.1250 ? [-1,-1,0]
Direction for atom Ir1[ 2] 0.7500 0.0000 0.6250 ? [-1,-1,0]
Direction for atom Ir1[ 3] 0.2500 0.0000 0.3750 ? [1,1,0]
Direction for atom Ir2[ 4] 0.7500 0.5000 0.3750 ? [1,-1,0]
Direction for atom Ir2[ 5] 0.2500 0.5000 0.6250 ? [-1,1,0]
Direction for atom Ir2[ 6] 0.2500 0.5000 0.1250 ? [-1,1,0]
Direction for atom Ir2[ 7] 0.7500 0.5000 0.8750 ? [1,-1,0]
Your input was [[1, 1, 0], [-1, -1, 0], [-1, -1, 0], [1, 1, 0], [1, -1, 0], [-1, 1, 0], [-1, 1, 0], [1, -1, 0]]. Is that OK [y/n]? y
Do you want short or long output [s/l]? s
Resuts written to output file moments.dat .
**** Correlated atom 1 ***** Mx_global= -0.90058469 0.00000000 0.00002456 -0.08717059 0.26840191 -0.00000071 0.00000317 1.19159160 0.00001365 -0.45228898 -0.61663031 0.00008483 0.86837991 -0.00009633 0.50136571 -0.00004848 -0.00000926 -0.57821711 0.00000307 0.33384331 0.00002456 0.08717059 0.90058469 0.00000000 0.00000316 -1.19159160 -0.26840191 -0.00000068 -0.61663031 -0.00008504 -0.00001394 -0.45228898 0.00000911 -0.57821711 -0.00000317 0.33384331 0.86837991 0.00009643 0.50136571 0.00004840 0.26840191 0.00000071 0.00000316 1.19159160 0.00002416 0.00000000 0.00010713 0.00001435 0.00000363 -0.01048982 0.08928393 0.00001147 1.01944113 0.00002073 0.58856989 -0.00001198 -0.00008313 0.67879375 0.00004800 -0.39189466 0.00000317 -1.19159160 -0.26840191 0.00000068 0.00010713 -0.00001435 -0.00002416 -0.00000000 0.08928393 -0.00001167 -0.00000331 -0.01048982 0.00008312 0.67879375 -0.00004798 -0.39189466 1.01944113 -0.00002076 0.58856989 0.00001197 0.00001365 0.45228898 -0.61663031 0.00008504 0.00000363 0.01048982 0.08928393 0.00001167 -0.02915968 0.00000000 0.00003726 0.10429920 0.00002930 0.07684550 0.00003117 0.35638077 0.63691047 0.00007845 0.10088571 -0.00000149 -0.61663031 -0.00008483 -0.00001394 0.45228898 0.08928393 -0.00001147 -0.00000331 0.01048982 0.00003726 -0.10429920 0.02915968 0.00000000 -0.63691047 0.00007841 -0.10088571 -0.00000166 0.00002896 -0.07684550 0.00003114 -0.35638077 0.86837991 0.00009633 0.00000911 0.57821711 1.01944113 -0.00002073 0.00008312 -0.67879375 0.00002930 -0.07684550 -0.63691047 -0.00007841 0.19667028 -0.00000000 0.00000000 -0.00000000 -0.00001058 0.98046968 -0.00000000 0.00000000 0.50136571 0.00004848 -0.00000317 -0.33384331 0.58856989 0.00001198 -0.00004798 0.39189466 0.00003117 -0.35638077 -0.10088571 0.00000166 0.00000000 0.00000000 0.19667028 0.00000000 0.00000000 0.00000000 -0.00001058 0.98046971 -0.00000926 0.57821711 0.86837991 -0.00009643 -0.00008313 -0.67879375 1.01944113 0.00002076 0.63691047 -0.00007845 0.00002896 0.07684550 -0.00001058 -0.98046968 -0.00000000 -0.00000000 -0.19667028 0.00000000 0.00000000 -0.00000000 0.00000307 -0.33384331 0.50136571 -0.00004840 0.00004800 0.39189466 0.58856989 -0.00001197 0.10088571 0.00000149 0.00003114 0.35638077 -0.00000000 -0.00000000 -0.00001058 -0.98046971 -0.00000000 0.00000000 -0.19667028 0.00000000 My_global= -0.08712554 -0.00000000 0.00004264 0.90061496 -1.19157653 0.00015728 0.00000425 0.26842503 -0.00013337 -0.61662147 0.45230117 -0.00002548 -0.57822320 0.00006427 -0.33382782 0.00006905 0.00000618 -0.86840643 0.00004103 0.50136837 0.00004264 -0.90061496 0.08712554 0.00000000 0.00000423 -0.26842503 1.19157653 0.00015730 0.45230117 0.00002525 0.00013356 -0.61662147 -0.00000605 -0.86840643 -0.00004098 0.50136837 -0.57822320 -0.00006407 -0.33382782 -0.00006915 -1.19157653 -0.00015728 0.00000423 0.26842503 -0.00001434 -0.00000000 0.00001265 0.00002416 -0.00000108 -0.08928620 -0.01047182 -0.00000071 -0.67878665 0.00003116 -0.39190477 -0.00001798 -0.00012486 1.01942511 0.00007208 -0.58857010 0.00000425 -0.26842503 1.19157653 -0.00015730 0.00001265 -0.00002416 0.00001434 0.00000000 -0.01047182 0.00000038 0.00000084 -0.08928620 0.00012482 1.01942511 -0.00007207 -0.58857010 -0.67878665 -0.00003115 -0.39190477 0.00001800 -0.00013337 0.61662147 0.45230117 -0.00002525 -0.00000108 0.08928620 -0.01047182 -0.00000038 -0.10429866 -0.00000000 0.00000319 -0.02916152 -0.00001242 -0.63690330 0.00006735 0.10087828 0.07683455 0.00003559 -0.35638215 -0.00004971 0.45230117 0.00002548 0.00013356 0.61662147 -0.01047182 0.00000071 0.00000084 0.08928620 0.00000319 0.02916152 0.10429866 -0.00000000 -0.07683455 0.00003589 0.35638215 -0.00004976 -0.00001251 0.63690330 0.00006755 -0.10087828 -0.57822320 -0.00006427 -0.00000605 0.86840643 -0.67878665 -0.00003116 0.00012482 -1.01942511 -0.00001242 0.63690330 -0.07683455 -0.00003589 -0.98046968 0.00000000 0.00000000 -0.00000000 -0.00004707 0.19667028 0.00000000 0.00000000 -0.33382782 -0.00006905 -0.00004098 -0.50136837 -0.39190477 0.00001798 -0.00007207 0.58857010 0.00006735 -0.10087828 0.35638215 0.00004976 -0.00000000 0.00000000 -0.98046971 -0.00000000 0.00000000 -0.00000000 -0.00004707 0.19667028 0.00000618 0.86840643 -0.57822320 0.00006407 -0.00012486 -1.01942511 -0.67878665 0.00003115 0.07683455 -0.00003559 -0.00001251 -0.63690330 -0.00004707 -0.19667028 0.00000000 -0.00000000 0.98046968 0.00000000 -0.00000000 -0.00000000 0.00004103 -0.50136837 -0.33382782 0.00006915 0.00007208 0.58857010 -0.39190477 -0.00001800 -0.35638215 0.00004971 0.00006755 0.10087828 0.00000000 -0.00000000 -0.00004707 -0.19667028 -0.00000000 0.00000000 0.98046971 -0.00000000 Mz_global= -0.00000140 0.00000000 -1.17689049 0.00009410 0.00008778 0.00000735 -0.00000216 0.00008393 -1.01731027 0.00010365 -0.00005365 -0.86999951 0.00001803 -0.00000000 0.00008655 0.68087923 -0.00000000 0.00004699 0.79616763 -0.00002609 -1.17689049 -0.00009410 0.00000227 0.00000000 -0.00000216 -0.00008398 -0.00008781 0.00000735 -0.00005395 0.86999951 1.01731027 0.00010333 0.00000000 0.00004699 -0.79616763 -0.00002569 0.00001803 0.00000000 0.00008695 -0.68087923 0.00008778 -0.00000735 -0.00000216 0.00008398 0.00000000 0.00000000 0.00000001 -0.00000000 -0.00001566 0.00004563 -0.00003863 0.00000119 0.00002116 -0.00000000 -0.00002460 0.00000235 0.00000001 -0.00005517 0.00001995 0.00002490 -0.00000216 -0.00008393 -0.00008781 -0.00000735 0.00000001 0.00000000 -0.00000000 0.00000000 -0.00003861 -0.00000119 0.00001566 0.00004561 -0.00000001 -0.00005517 -0.00001995 0.00002488 0.00002116 0.00000000 -0.00002462 -0.00000235 -1.01731027 -0.00010365 -0.00005395 -0.86999951 -0.00001566 -0.00004563 -0.00003861 0.00000119 0.00000364 0.00000000 -0.17689049 0.00005696 0.00000000 0.00002917 1.70367860 -0.00000852 -0.00001036 -0.00000000 0.00001588 0.00000000 -0.00005365 0.86999951 1.01731027 -0.00010333 -0.00003863 -0.00000119 0.00001566 -0.00004561 -0.17689049 -0.00005696 -0.00000450 -0.00000000 0.00001036 -0.00000000 -0.00001554 0.00000000 0.00000000 -0.00002917 1.70367860 0.00000852 0.00001803 0.00000000 0.00000000 -0.00004699 0.00002116 0.00000000 -0.00000001 0.00005517 0.00000000 -0.00002917 0.00001036 0.00000000 0.00004407 0.00000000 -0.00000000 0.00000000 -1.00000000 -0.00001963 0.00000000 0.00000000 0.00008655 -0.68087923 -0.79616763 0.00002569 -0.00002460 -0.00000235 -0.00001995 -0.00002488 1.70367860 0.00000852 -0.00001554 -0.00000000 -0.00000000 0.00000000 0.00004407 0.00000000 -0.00000000 0.00000000 -1.00000002 -0.00001963 -0.00000000 -0.00004699 0.00001803 -0.00000000 0.00000001 0.00005517 0.00002116 -0.00000000 -0.00001036 0.00000000 0.00000000 0.00002917 -1.00000000 0.00001963 -0.00000000 -0.00000000 -0.00004407 0.00000000 0.00000000 0.00000000 0.79616763 0.00002609 0.00008695 0.68087923 0.00001995 -0.00002490 -0.00002462 0.00000235 0.00001588 -0.00000000 1.70367860 -0.00000852 0.00000000 -0.00000000 -1.00000002 0.00001963 0.00000000 -0.00000000 -0.00004407 0.00000000
**** Correlated atom 1 ***** New CF Transformation= 0.08108551 0.24904873 -0.00002233 -0.00001854 0.00000000 0.00000000 -0.23053569 0.55647504 -0.08108551 -0.24904873 0.08109960 -0.24901336 0.23047641 0.55650662 0.00000000 0.00000000 -0.00003701 0.00001293 -0.08109960 0.24901336 -0.08109954 -0.24901322 0.00003699 0.00001293 0.00000000 -0.00000000 -0.23047636 0.55650651 0.08109954 0.24901322 0.08108557 -0.24904886 -0.23053573 -0.55647515 0.00000000 -0.00000000 -0.00002232 0.00001854 -0.08108557 0.24904886 0.00000055 -0.00000434 -0.27056219 0.65330804 0.00000000 0.00000000 -0.00003297 -0.00002241 -0.00000055 0.00000434 -0.00000328 -0.00001400 -0.00003878 0.00003069 0.00000000 0.00000000 0.27063392 0.65325494 0.00000328 0.00001400 0.00000328 -0.00001401 -0.27063392 0.65325494 0.00000000 0.00000000 0.00003876 0.00003069 -0.00000328 0.00001401 0.00000055 0.00000434 -0.00003295 0.00002241 0.00000000 0.00000000 -0.27056219 -0.65330804 -0.00000055 -0.00000434 -0.16299241 0.39349848 -0.00000091 0.00003600 0.00000000 0.00000000 0.11470054 0.35217760 0.16299241 -0.39349848 0.16299238 0.39349840 0.11466384 -0.35218854 0.00000000 0.00000000 0.00000099 0.00002844 -0.16299238 -0.39349840 -0.16299241 0.39349848 -0.00000097 0.00002845 0.00000000 0.00000000 -0.11466392 -0.35218872 0.16299241 -0.39349848 -0.16299238 -0.39349840 0.11470047 -0.35217743 0.00000000 0.00000000 -0.00000089 -0.00003601 0.16299238 0.39349840 0.00000000 0.00000000 0.00000000 0.00000000 -0.27059805 0.65328148 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.27059805 0.65328148 0.00000000 0.00000000 0.00000000 0.00000000 -0.19134172 0.46193977 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.19134172 0.46193977 0.19134172 0.46193977 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.19134172 0.46193977 0.00000000 0.00000000 0.00000000 0.00000000 -0.27059805 0.65328148 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.27059805 -0.65328148 0.00000000 0.00000000 0.00000000 0.00000000 -0.19134172 0.46193977 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.19134172 0.46193977 -0.19134172 -0.46193977 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.19134172 -0.46193977
To allow for the broken symmetry state, we also need to change the Sigind matrix in Sr2IrO4.indmfl file. For the first atom, the entry in Sr2IrO4.indmfl file should finally take the form:
#================ # Siginds and crystal-field transformations for correlated orbitals ================ 8 10 6 # Number of independent kcix blocks, max dimension, max num-independent-components 1 10 6 # cix-num, dimension, num-independent-components #---------------- # Independent components are -------------- 'xz' 'yz' 'xy' 'xz' 'yz' 'xy' #---------------- # Sigind follows -------------------------- 1 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 4 0 0 0 0 0 0 0 0 0 0 5 0 0 0 0 0 0 0 0 0 0 6 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 #---------------- # Transformation matrix follows ----------- 0.08108551 0.24904873 -0.00002233 -0.00001854 0.00000000 0.00000000 -0.23053569 0.55647504 -0.08108551 -0.24904873 0.08109960 -0.24901336 0.23047641 0.55650662 0.00000000 0.00000000 -0.00003701 0.00001293 -0.08109960 0.24901336 -0.08109954 -0.24901322 0.00003699 0.00001293 0.00000000 -0.00000000 -0.23047636 0.55650651 0.08109954 0.24901322 0.08108557 -0.24904886 -0.23053573 -0.55647515 0.00000000 -0.00000000 -0.00002232 0.00001854 -0.08108557 0.24904886 0.00000055 -0.00000434 -0.27056219 0.65330804 0.00000000 0.00000000 -0.00003297 -0.00002241 -0.00000055 0.00000434 -0.00000328 -0.00001400 -0.00003878 0.00003069 0.00000000 0.00000000 0.27063392 0.65325494 0.00000328 0.00001400 0.00000328 -0.00001401 -0.27063392 0.65325494 0.00000000 0.00000000 0.00003876 0.00003069 -0.00000328 0.00001401 0.00000055 0.00000434 -0.00003295 0.00002241 0.00000000 0.00000000 -0.27056219 -0.65330804 -0.00000055 -0.00000434 -0.16299241 0.39349848 -0.00000091 0.00003600 0.00000000 0.00000000 0.11470054 0.35217760 0.16299241 -0.39349848 0.16299238 0.39349840 0.11466384 -0.35218854 0.00000000 0.00000000 0.00000099 0.00002844 -0.16299238 -0.39349840 -0.16299241 0.39349848 -0.00000097 0.00002845 0.00000000 0.00000000 -0.11466392 -0.35218872 0.16299241 -0.39349848 -0.16299238 -0.39349840 0.11470047 -0.35217743 0.00000000 0.00000000 -0.00000089 -0.00003601 0.16299238 0.39349840 0.00000000 0.00000000 0.00000000 0.00000000 -0.27059805 0.65328148 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.27059805 0.65328148 0.00000000 0.00000000 0.00000000 0.00000000 -0.19134172 0.46193977 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.19134172 0.46193977 0.19134172 0.46193977 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.19134172 0.46193977 0.00000000 0.00000000 0.00000000 0.00000000 -0.27059805 0.65328148 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.27059805 -0.65328148 0.00000000 0.00000000 0.00000000 0.00000000 -0.19134172 0.46193977 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.19134172 0.46193977 -0.19134172 -0.46193977 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 -0.19134172 -0.46193977
dmft_copy.py <dir-with-LDA-results>
szero.py
# s_oo= [18.65, 18.65, 18.65, 18.65, 18.65, 18.65] # Edc= [18.65, 18.65, 18.65, 18.65, 18.65, 18.65]
# s_oo= [19.65, 17.65, 18.65, 18.65, 18.65, 18.65] # Edc= [18.65, 18.65, 18.65, 18.65, 18.65, 18.65]
Now we are ready to submit the job. Since both DFT and DMFT steps are slow, we should execute them both in parallel, which was explained in previous tutorials.