[1] Electronic structure calculations with dynamical
mean-field theory, G. Kotliar, S. Y. Savrasov, K. Haule,
et.al., Rev. Mod. Phys. 78, 865 (2006).
[2] Dynamical mean-field theory within the full-potential
methods, K. Haule, C.-H. Yee, and K. Kim, Phys. Rev. B 81, 195107 (2010).
[3] Exact double-counting in combining the Dynamical Mean
Field Theory and the Density Functional Theory, K. Haule,
PRL 115, 196403 (2015).
[4] Quantum Monte Carlo Impurity Solver for Cluster DMFT and
Electronic Structure Calculations, Kristjan Haule,
Phys. Rev. B 75, 155113 (2007).
[5] Free energy from stationary implementation of the DFT+DMFT
functional, K. Haule and T. Birol,
PRL 115, 256402 (2015).
[6] Forces for Structural Optimizations in Correlated
Materials within DFT+Embedded DMFT Functional Approach, K. Haule,
G. L. Pascut,
arXiv:1602.02819.
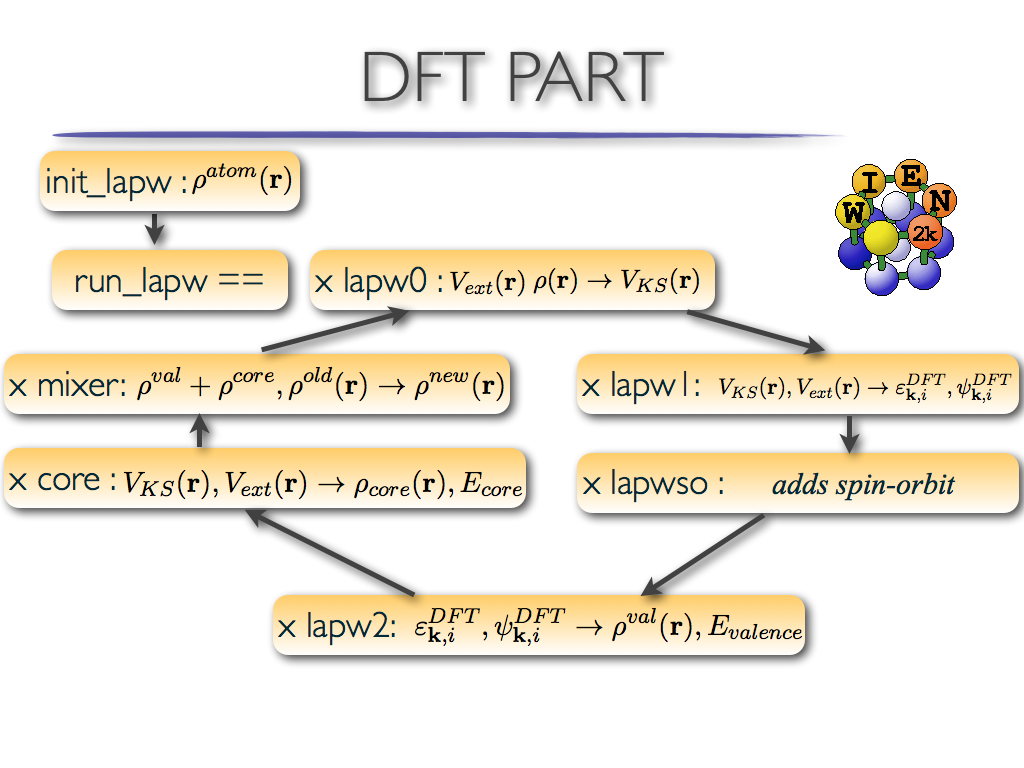
The Density Functional Theory implementation within Wien2k consists of
several steps
(see wien2k-documentation)
depicted in the Figure:
The steps are:
lapw0: calculates electronic potential
lapw1: solves eigenvalue problem for Kohn-Sham orbitals
lapwso: adds spin-orbit coupling
lapw2: calculates DFT valence electronic charge
core: adds core states to DFT charge
mixer: mixes electronic charge density
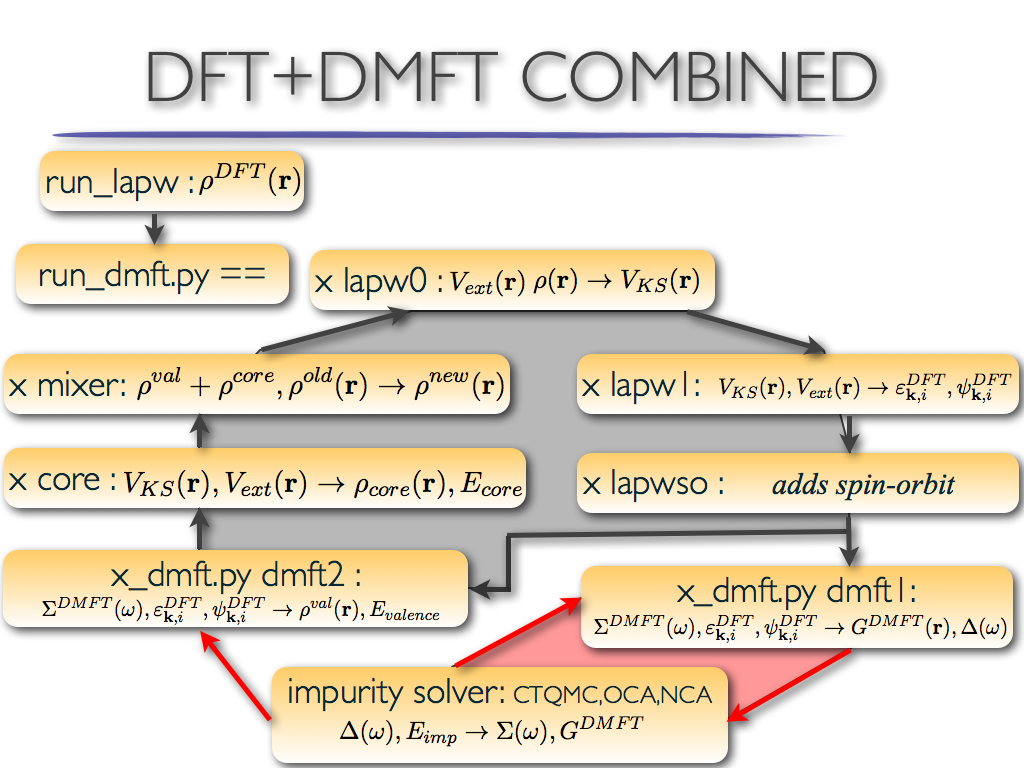
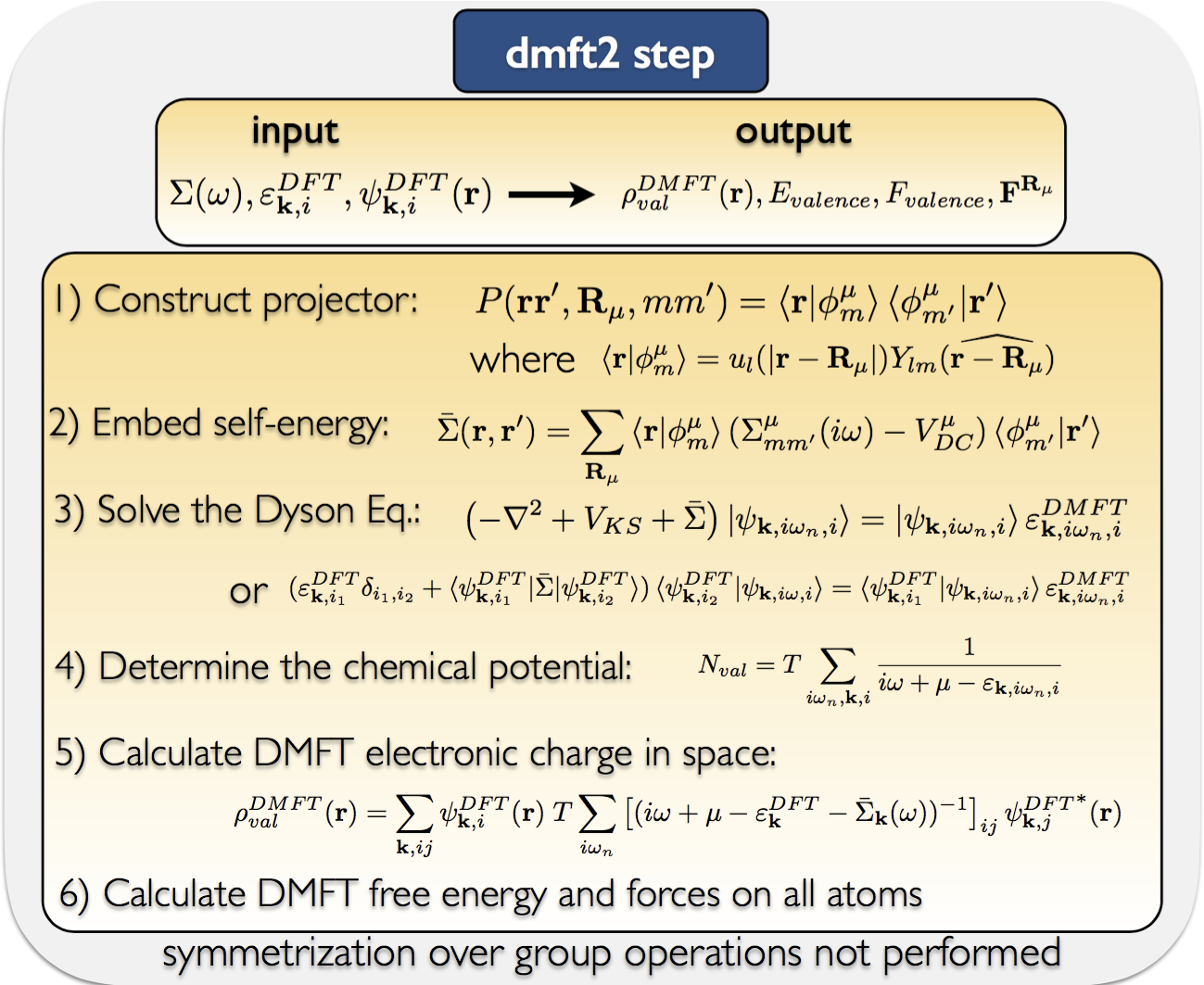
The DMFT+DMFT inserts two new steps (dmft1 and impurity solver) and
replaces lapw2 with dmft2 step. The process looks like that:
The eDMFT steps are:
lapw0: calculates electronic potential
lapw1: solves eigenvalue problem for Kohn-Sham orbitals
lapwso: adds spin-orbit coupling
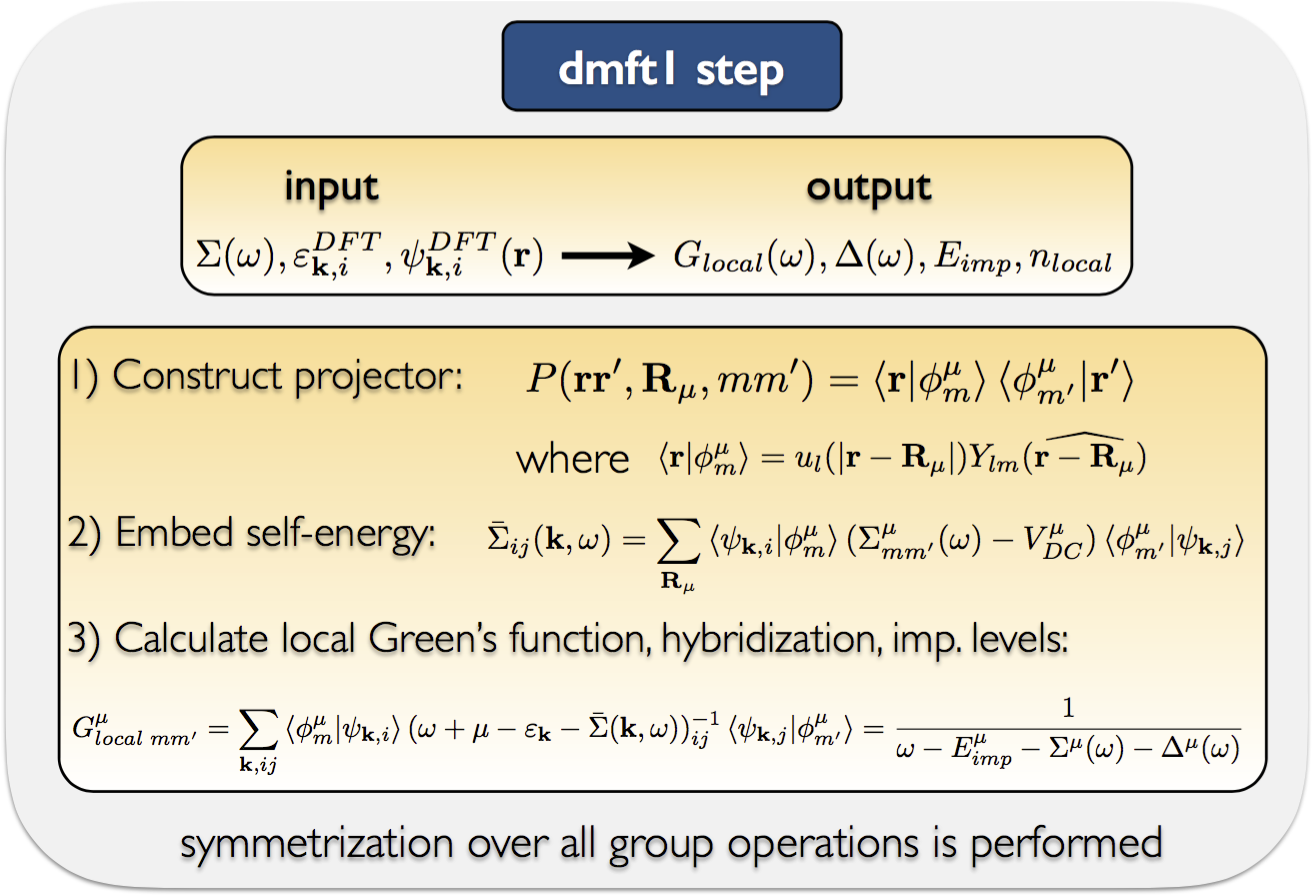
dmft1: calculates local green's function and hybridization function
impurity: solves the auxiliary impurity problem for electronic self-energy correction
The eDMFT can be broaken into two loops: the DFT loop (grey area in
Figure above) and the DMFT loop (red area in Figure above). We can
repeat several times the DMFT loop to better converge the DMFT self-energy
computed by the impurity solver (this part is so-called non-charge
self-consistent DMFT). The DFT charge loop can also be iterated
several times using fixed self-energy to better converge electronic
charge.
The number of DFT steps and DMFT steps can be controled by the input
file "params.dat". The fastest convergence is usually achieved by
requiring that roughly the same amount of time is spend in both steps.