
45 156 1 5 # hybridization band index nemin and nemax, renormalize for interstitials, projection type 0 0.025 0.025 200 -3.000000 1.000000 # matsubara, broadening-corr, broadening-noncorr, nomega, omega_min, omega_max (in eV) 4 # number of correlated atoms 1 1 0 # iatom, nL, locrot 2 3 1 # L, qsplit, cix 2 1 0 # iatom, nL, locrot 2 3 2 # L, qsplit, cix 3 1 0 # iatom, nL, locrot 2 3 3 # L, qsplit, cix 4 1 0 # iatom, nL, locrot 2 3 4 # L, qsplit, cix
The second line contains a switch for the real-axis or imaginary axis calculation. For ctqmc impurity solver, we need to set "matsubara" to 1. The next two numbers specify the broadening in k-point summation. The last three numbers are used for plotting the spectral functions, to which we will return latter.
The third line specifies the number of correlated atoms, which is here 4, i.e., all V-atoms in the unit cell.
The next eigth lines can be grouped into set of 4 x 2, i.e., for each correlated V atom two lines. The first line in the group specifies the consequitive atom number (from case.struct), which we treat as correlated. It also specifies how many orbital indeces (different L's) we will use for projection (only 1). The last index "locrot" is used for rotation of local axis, and will be used it shortly, to specify the local coordinate system for DMFT basis.
The second line in a group specifies orbital momentum "l=2",
"qsplit=3" which we choose during initialization. Each correlated
orbital-set gets a unique index (correlated index) during
initialization. These 4 blocks are enumerated by consecutive numbers
cix=1,2,3,4. For each of these 4 correlated sets, a more detailed
specification is given in the remaining of "case.indmfl" file.
The global axis of the structure is not convenient for DMFT
calculation. While in principle the DMFT is rotationally invariant
method, and any axis should give the same result, in practice there is
enormous difference in the severity of the quamtum Monte Carlo sign
problem. To reduce/avoid sign problem, we always pick the local axis on
each atom so that the hybridization matrix is maximally diagonal. This
is usually the local axis, which is aligned with the local environment of
the correlated atom, in this case octahedron. In this figure, we show
how the local axis on V1 will be choosen below.
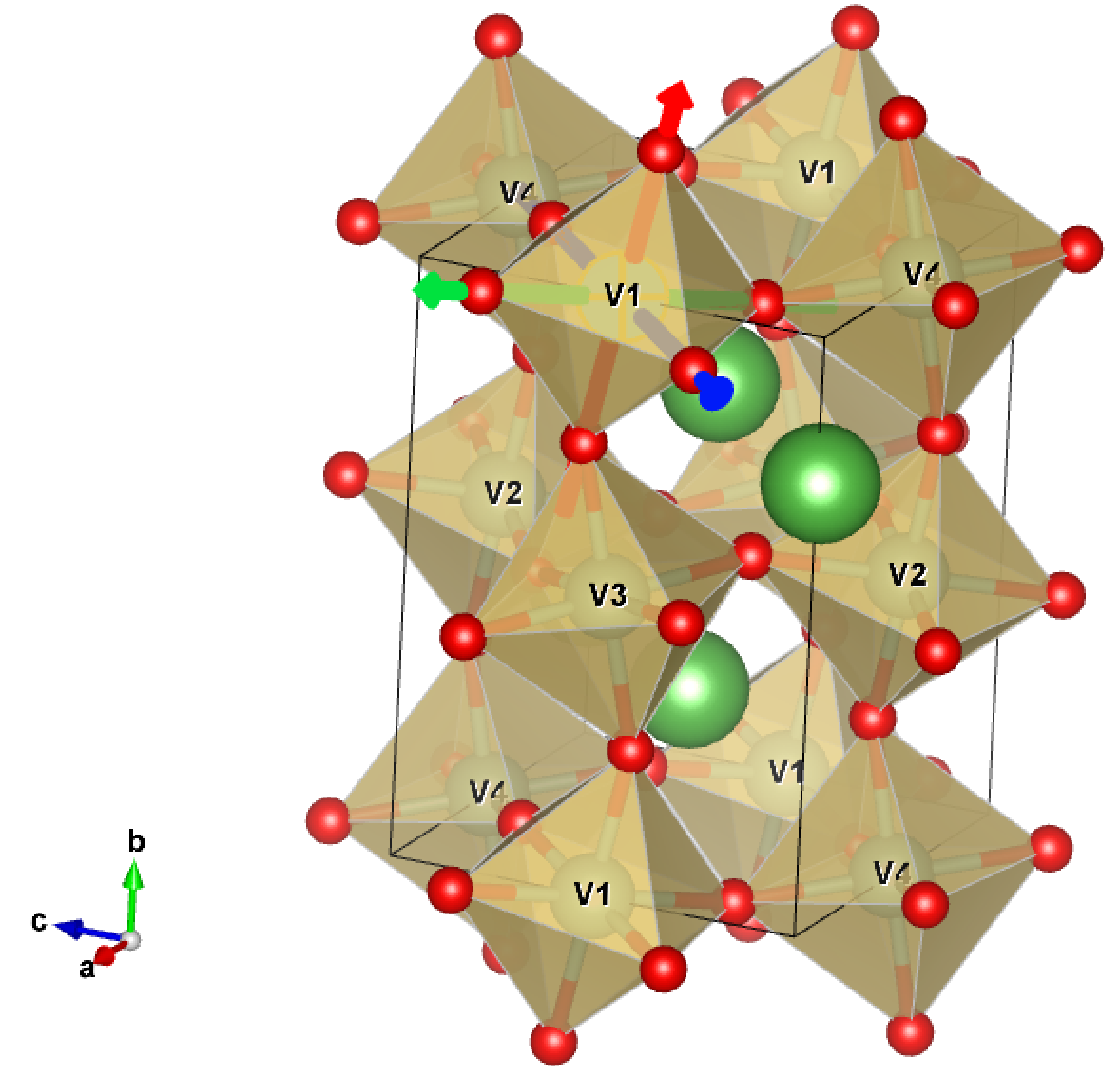
localaxes.py
1 ATOM: 1 EQUIV. 1 V 3+ AT 0.00000 0.00000 0.50000 2 ATOM: 1 EQUIV. 2 V 3+ AT 0.50000 0.50000 0.00000 3 ATOM: 1 EQUIV. 3 V 3+ AT 0.00000 0.50000 0.50000 4 ATOM: 1 EQUIV. 4 V 3+ AT 0.50000 0.00000 0.00000 ...... Enter the atom for which you want to determine local axes: 1
trying cube :-N= -2.000045418 chi2= 0.0781099540504 = 3.78604380855 combined-criteria 5.54201036661 trying octahedra : -N= -4.55779706225e-05 chi2= 0.000198898591793 = 3.78604379544 combined-criteria 0.00392610291121 trying tetrahedra : -N= -2.31787341618e-05 chi2= 0.582093832999 = 3.78514737663 combined-criteria 11.4900717126 trying square-piramid : -N= -3.66084575019e-05 chi2= 0.493633610179 = 3.78568492083 combined-criteria 9.74393690443 According to angle variance and bond-angle we are trying octahedra with = 4.55779706225e-05 and = 0.000198898591793 Found the environment is ('octahedra', 4.5577970622545649e-05, 0.00019889859179304857) Rotation to input into case.indmfl by locrot=-1 : 0.79186901 0.14777288 0.59254253 -0.03062084 0.97866938 -0.20314676 -0.60992282 0.14272147 0.77950288
Let us first understand this transformation matrix. It is a unitary transformation from global cartesian coordinate system to local cartesian coordinate system. New x-axis, y-axis and z-axis are given in the first, the second and the third row. One can plot these vectors in VESTA to visualize our choice. The matrix is not written in lattice coordinates, but cartesian coordinates. If one wants the same transformation in lattice coordinates, one needs to build a transformation from cartesian to lattice coordinates. There are several ways to do that. One can look at LaVO3.outputkgen, and find R1,R2,R3 at the top of the file
R1 = 10.498336 0.000000 0.000000 R2 = 0.000000 14.831856 0.000000 R3 = 0.000000 0.000000 10.494575
The same information exists also in LaVO3.outputd under BR1_DIR. The transformation from cartesian to lattice coordinates is: C2L = Inverse([[R1x,R2x,R3x],[R1y,R2y,R3y],[R1z,R2z,R3z]]) which is here
0.0952532 0.0000000 0.0000000 0.0000000 0.0674224 0.0000000 0.0000000 0.0000000 0.0952873
hence new x-axis in lattice coordinates is C2L*(new-x) = (-0.00291502, 0.0659845, -0.0193558), etc. An alternative way to find transformation from cartesian to lattice coordinates is to inspect case.rotl (which appears after dmft1 is being run) at the top of the file one finds
BR1 0.59849 0.00000 0.00000 -0.00000 0.42363 0.00000 -0.00000 -0.00000 0.59871
It turns out that C2L = BR1/(2*pi).
Now that we have the transformation matrix to local coordinate system, we will add it to LaVO3.indmfl file. We will change locrot to -1 for the relevant atom, and paste below the transformation for that atom. The result for the first atom looks like that
1 1 -1 # iatom, nL, locrot 2 3 1 # L, qsplit, cix 0.79186901 0.14777288 0.59254253 -0.03062084 0.97866938 -0.20314676 -0.60992282 0.14272147 0.77950288
The header of the LaVO3.struct file finally looks like that
45 156 1 5 # hybridization band index nemin and nemax, renormalize for interstitials, projection type 0 0.025 0.025 200 -3.000000 1.000000 # matsubara, broadening-corr, broadening-noncorr, nomega, omega_min, omega_max (in eV) 4 # number of correlated atoms 1 1 -1 # iatom, nL, locrot 2 3 1 # L, qsplit, cix 0.79186901 0.14777288 0.59254253 -0.03062084 0.97866938 -0.20314676 -0.60992282 0.14272147 0.77950288 2 1 -1 # iatom, nL, locrot 2 3 2 # L, qsplit, cix 0.79186901 0.14777288 0.59254253 -0.03062084 0.97866938 -0.20314676 -0.60992282 0.14272147 0.77950288 3 1 -1 # iatom, nL, locrot 2 3 3 # L, qsplit, cix 0.79186901 0.14777288 0.59254253 -0.03062084 0.97866938 -0.20314676 -0.60992282 0.14272147 0.77950288 4 1 -1 # iatom, nL, locrot 2 3 4 # L, qsplit, cix 0.79186901 0.14777288 0.59254253 -0.03062084 0.97866938 -0.20314676 -0.60992282 0.14272147 0.77950288
The specification of correlated set is given in the second part of case.indmfl, which we did not change.
We next create a blank self-energy file to plot the Green's function in the new local coordinate system. We just execute
szero.py
Next we will run
x_dmft.py lapw1 x_dmft.py dmft1
If you wish to run these runs in parallel, you just need to specify mpi_prefix.dat file, which must contain the mpi_run command.
We can examine LaVO3.gc[1-4] which should all contain almost identical Green's function, which proves that the transformation between the four equivalent atoms was properly chosen. Next we realize that there is a splitting between the t2g orbitals, and we will choose a local transformation, which will make hybridization as diagonal as possible. To do that, we first examine LaVO3.outputdmf1 file, which contains (towards the end) the following lines
Full matrix of impurity levels follows
icix= 1
-0.01624121 0.00000000 -0.00712563 0.00000000 -0.01627386 0.00000000
-0.00712563 -0.00000000 -0.01061912 -0.00000000 0.01733578 -0.00000000
-0.01627386 -0.00000000 0.01733578 0.00000000 -0.02757288 0.00000000
icix= 1 at omega=0
0.24430529 0.00000000 0.01913794 0.00000000 0.00245632 0.00000000
0.01913794 -0.00000000 0.26547163 -0.00000000 -0.03205887 0.00000000
0.00245632 -0.00000000 -0.03205887 -0.00000000 0.25124493 0.00000000
Obtaining transformation by findRot.py
When using this script, we just typefindRot.py
x_dmft.py dmft1
Obtaining transformation by a series of steps
We first prepare a text file, which contains the matrix of impurity levels from LaVO3.outputdmf1, and also the current transformation from spherical to cubic harmonics, which is specified in LaVO3.indmfl file. We will create a python text rot.dat file, which will contain the impurity levels in strHc variable and the transformation in strT2C variable:strHc=""" 0.24430529 -0.00000000 0.01913794 0.00000000 0.00245632 -0.00000000 0.01913794 -0.00000000 0.26547163 -0.00000000 -0.03205887 0.00000000 0.00245632 0.00000000 -0.03205887 -0.00000000 0.25124493 -0.00000000 """ strT2C=""" 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 -0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 -0.00000000 -0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 """
find3dRotation.py rot.dat
:SUCCESS For orbital [0] the final transformation is real :SUCCESS For orbital [1] the final transformation is real :SUCCESS For orbital [2] the final transformation is real Rotation to input : 0.00000000 0.43702058 0.34959590 -0.43219872 0.00000000 0.00000000 -0.34959590 -0.43219872 0.00000000 -0.43702058 0.00000000 -0.39025811 0.58445888 0.07814369 0.00000000 0.00000000 -0.58445888 0.07814369 0.00000000 0.39025811 0.00000000 -0.39586820 -0.19023811 -0.55416409 0.00000000 0.00000000 0.19023811 -0.55416409 0.00000000 0.39586820
After the replacement of transformation matrices, the file LaVO3.indmfl contains:
#================ # Siginds and crystal-field transformations for correlated orbitals ================ 4 5 3 # Number of independent kcix blocks, max dimension, max num-independent-components 1 5 3 # cix-num, dimension, num-independent-components #---------------- # Independent components are -------------- 'xz' 'yz' 'xy' #---------------- # Sigind follows -------------------------- 1 0 0 0 0 0 2 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 0 0 #---------------- # Transformation matrix follows ----------- 0.00000000 0.43702058 0.34959590 -0.43219872 0.00000000 0.00000000 -0.34959590 -0.43219872 0.00000000 -0.43702058 0.00000000 -0.39025811 0.58445888 0.07814369 0.00000000 0.00000000 -0.58445888 0.07814369 0.00000000 0.39025811 0.00000000 -0.39586820 -0.19023811 -0.55416409 0.00000000 0.00000000 0.19023811 -0.55416409 0.00000000 0.39586820 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 2 5 3 # cix-num, dimension, num-independent-components #---------------- # Independent components are -------------- 'xz' 'yz' 'xy' #---------------- # Sigind follows -------------------------- 1 0 0 0 0 0 2 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 0 0 #---------------- # Transformation matrix follows ----------- 0.00000000 0.43702058 0.34959590 -0.43219872 0.00000000 0.00000000 -0.34959590 -0.43219872 0.00000000 -0.43702058 0.00000000 -0.39025811 0.58445888 0.07814369 0.00000000 0.00000000 -0.58445888 0.07814369 0.00000000 0.39025811 0.00000000 -0.39586820 -0.19023811 -0.55416409 0.00000000 0.00000000 0.19023811 -0.55416409 0.00000000 0.39586820 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 3 5 3 # cix-num, dimension, num-independent-components #---------------- # Independent components are -------------- 'xz' 'yz' 'xy' #---------------- # Sigind follows -------------------------- 1 0 0 0 0 0 2 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 0 0 #---------------- # Transformation matrix follows ----------- 0.00000000 0.43702058 0.34959590 -0.43219872 0.00000000 0.00000000 -0.34959590 -0.43219872 0.00000000 -0.43702058 0.00000000 -0.39025811 0.58445888 0.07814369 0.00000000 0.00000000 -0.58445888 0.07814369 0.00000000 0.39025811 0.00000000 -0.39586820 -0.19023811 -0.55416409 0.00000000 0.00000000 0.19023811 -0.55416409 0.00000000 0.39586820 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 4 5 3 # cix-num, dimension, num-independent-components #---------------- # Independent components are -------------- 'xz' 'yz' 'xy' #---------------- # Sigind follows -------------------------- 1 0 0 0 0 0 2 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 0 0 #---------------- # Transformation matrix follows ----------- 0.00000000 0.43702058 0.34959590 -0.43219872 0.00000000 0.00000000 -0.34959590 -0.43219872 0.00000000 -0.43702058 0.00000000 -0.39025811 0.58445888 0.07814369 0.00000000 0.00000000 -0.58445888 0.07814369 0.00000000 0.39025811 0.00000000 -0.39586820 -0.19023811 -0.55416409 0.00000000 0.00000000 0.19023811 -0.55416409 0.00000000 0.39586820 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000
We now rerun the dmft1 step
x_dmft.py dmft1
Full matrix of impurity levels follows
icix= 1
0.00888017 -0.00000000 0.00353679 0.00000000 0.00345745 0.00000000
0.00353679 -0.00000000 -0.03365887 0.00000000 -0.01070044 -0.00000000
0.00345745 -0.00000000 -0.01070044 0.00000000 -0.02965450 0.00000000
icix= 1 at omega=0
0.21757484 -0.00000000 -0.00000000 -0.00000000 -0.00000000 -0.00000000
-0.00000000 0.00000000 0.24850424 0.00000000 -0.00000000 -0.00000000
-0.00000000 0.00000000 -0.00000000 0.00000000 0.29494278 0.00000000
The rest of initialization
Finally, DMFT initialization produced a second file, named case.indmfi, which contains the following lines:1 # number of sigind blocks 5 # dimension of this sigind block 1 0 0 0 0 0 2 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 0 0
Next create a new folder, and when in that folder, use the command
dmft_copy.py <lda_results_directory>
There are a few more steps we need to to:
- Next, edit LaVO3.indmfl and change the flag matsubara in second line from 0 to 1, because the impurity solver works on imaginary axis.
- delete sig.inp file, which is given on real axis.
- run szero.py to create a blank self-energy on imaginary axis.
- copy params.dat file into working
directory, which contains
and is basically the same as in previous tutorials. The only difference is that nominal valence is here 2.0, and we will not recompute the chemical potential, as we expect a Mott-gap. We need to be careful that the solid is neutral (charge density is correct) by examining :DRHO in LaVO3.scf or the tail of dmft2_info.out. Note that at the beginning, some mismatch of charge is acceptable, but not once the charge is converged, :DRHO should be very small.
solver = 'CTQMC' # impurity solver DCs = 'nominal' # double counting scheme max_dmft_iterations = 3 # number of iteration of the dmft-loop only max_lda_iterations = 100 # number of iteration of the LDA-loop only finish = 30 # number of iterations of full charge loop (1 = no charge self-consistency) ntail = 300 # on imaginary axis, number of points in the tail of the logarithmic mesh cc = 5e-5 # the charge density precision to stop the LDA+DMFT run ec = 5e-5 # the energy precision to stop the LDA+DMFT run recomputeEF = 0 # Recompute EF in dmft2 step. # Impurity problem number 0 iparams0={"exe" : ["ctqmc" , "# Name of the executable"], "U" : [6.0 , "# Coulomb repulsion (F0)"], "J" : [0.8 , "# Coulomb repulsion (F0)"], "beta" : [50 , "# Inverse temperature"], "CoulombF" : ["'Ising'" , "# Form of Coulomb repulsion"], "svd_lmax" : [25 , "# We will use SVD basis to expand G, with this cutoff"], "M" : [10e6 , "# Total number of Monte Carlo steps"], "mode" : ["SH" , "# We will use self-energy sampling, and Hubbard I tail"], "nom" : [200 , "# Number of Matsubara frequency points sampled"], "tsample" : [200 , "# How often to record measurements"], "GlobalFlip" : [1000000 , "# How often to try a global flip"], "warmup" : [1e6 , "# Warmup number of QMC steps"], "nf0" : [2 , "# Double counting parameter"], }We notice that DCs='exactd' gives almost identical results. It is not surprising that dielectric screening is most appropriate for insulator like LaVO3.
Now we are ready to run DFT+DMFT calculation.
As in previous tutorial, you should set-up mpi_prefix.dat,
which contains the command for running mpi job on your computer, for
example
echo "mpirun -n $NSLOTS" > mpi_prefix.dat
or
echo "mpiexec -port $port -np $NSLOTS -machinefile $TMPDIR/machines" > mpi_prefix.dat,
depending on your system.
Then call the script "$WIEN_DMFT_ROOT/run_dmft.py".
Make sure that the total number of Monte Carlo steps is not too
small. For example, M=20e6 on 60 cores should give reasonable
precision. If you have only 10 cores, you should increase M to 120e6.
Again, you should check that the system is properly set-up, i.e., that
the computing notes have the
following environmental
variable specified: WIENROOT, WIEN_DMFT_ROOT, SCRATCH. Also make sure
that $WIENROOT and $WIEN_DMFT_ROOT are in $PATH as well as
$WIEN_DMFT_ROOT is in $PYTHONPATH. Typically, the .bashrc will contain
the following lines:
export WIENROOT=< wien-instalation-dir > export WIEN_DMFT_ROOT=< dir-containing-dmft-executables > export SCRATCH="." export PATH=$PATH:$WIENROOT:$WIEN_DMFT_ROOT export PYTHONPATH=$PYTHONPATH:$WIEN_DMFT_ROOT
During the run, we will monitor information file and update parameters, if necessary. The ':log' file lists the individual modules that are running:
Sat Mar 25 23:54:05 EDT 2017> (x) -f LaVO3 lapw0 Sat Mar 25 23:54:19 EDT 2017> lapw1 Sat Mar 25 23:54:29 EDT 2017> dmft1 Sat Mar 25 23:54:34 EDT 2017> impurity Sat Mar 25 23:56:17 EDT 2017> dmft1 Sat Mar 25 23:56:26 EDT 2017> impurity Sat Mar 25 23:58:10 EDT 2017> dmft1 Sat Mar 25 23:58:15 EDT 2017> impurity Sat Mar 25 23:59:50 EDT 2017> dmft2 Sun Mar 26 00:00:11 EDT 2017> (x) -f LaVO3 lcore Sun Mar 26 00:00:12 EDT 2017> (x) -f LaVO3 mixer Sun Mar 26 00:00:23 EDT 2017> (x) -f LaVO3 lapw0 Sun Mar 26 00:00:37 EDT 2017> lapw1 Sun Mar 26 00:00:47 EDT 2017> dmft2 Sun Mar 26 00:01:08 EDT 2017> (x) -f LaVO3 lcore Sun Mar 26 00:01:09 EDT 2017> (x) -f LaVO3 mixer Sun Mar 26 00:01:20 EDT 2017> (x) -f LaVO3 lapw0 Sun Mar 26 00:01:34 EDT 2017> lapw1 Sun Mar 26 00:01:43 EDT 2017> dmft2 Sun Mar 26 00:02:05 EDT 2017> (x) -f LaVO3 lcore Sun Mar 26 00:02:06 EDT 2017> (x) -f LaVO3 mixer Sun Mar 26 00:02:17 EDT 2017> (x) -f LaVO3 lapw0 Sun Mar 26 00:02:31 EDT 2017> lapw1 Sun Mar 26 00:02:40 EDT 2017> dmft2 Sun Mar 26 00:03:02 EDT 2017> (x) -f LaVO3 lcore Sun Mar 26 00:03:03 EDT 2017> (x) -f LaVO3 mixer Sun Mar 26 00:03:13 EDT 2017> (x) -f LaVO3 lapw0 Sun Mar 26 00:03:27 EDT 2017> lapw1 Sun Mar 26 00:03:37 EDT 2017> dmft2 Sun Mar 26 00:03:59 EDT 2017> (x) -f LaVO3 lcore Sun Mar 26 00:04:00 EDT 2017> (x) -f LaVO3 mixer Sun Mar 26 00:04:10 EDT 2017> (x) -f LaVO3 lapw0 Sun Mar 26 00:04:24 EDT 2017> lapw1 Sun Mar 26 00:06:29 EDT 2017> dmft2 Sun Mar 26 00:07:10 EDT 2017> (x) -f LaVO3 lcore Sun Mar 26 00:07:11 EDT 2017> (x) -f LaVO3 mixer Sun Mar 26 00:07:22 EDT 2017> (x) -f LaVO3 lapw0
We can check the convergence of the charge by "grep ':CHARGE' LaVO3.dayfile" and plotting the result shows
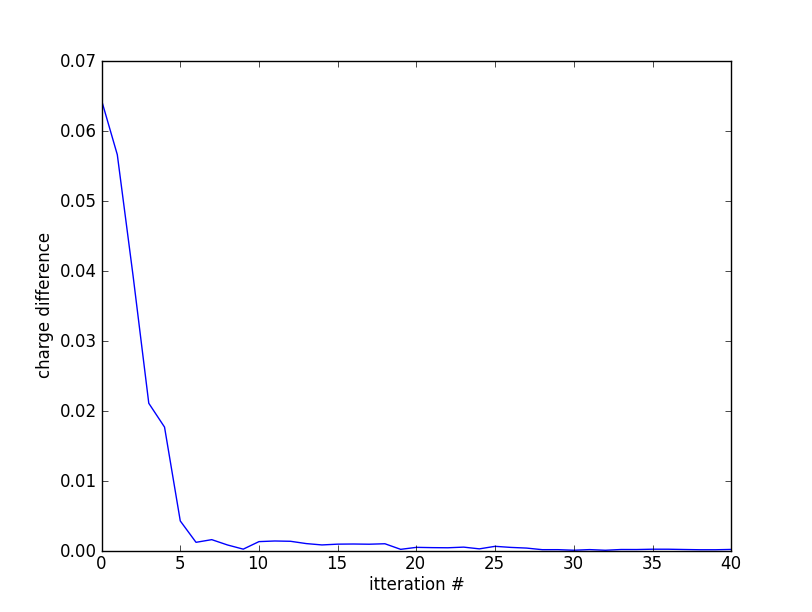
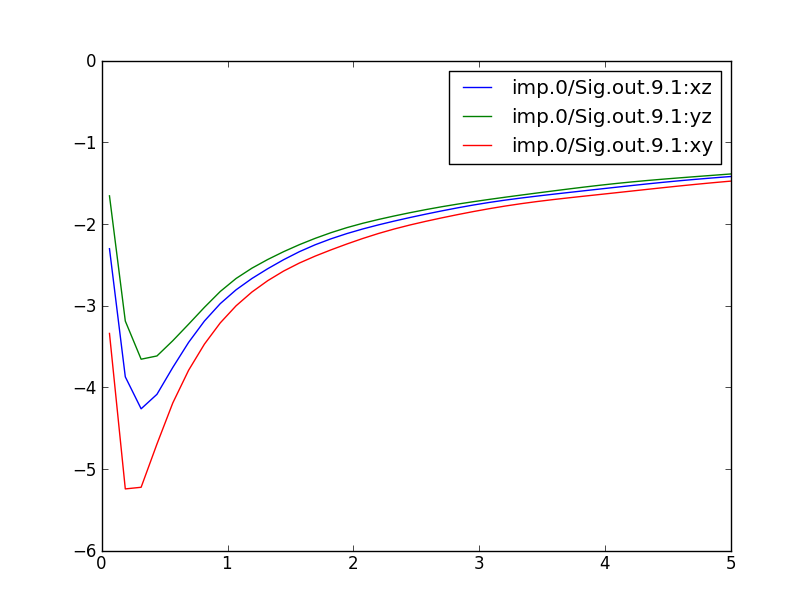
Another important file to check is the info.iterate, which contains information about the chemical potential, the number of electrons on the V ion, etc:
# #. # mu Vdc Etot Ftot+T*Simp Ftot+T*Simp n_latt n_imp Eimp[0] Eimp[-1] 0 0. 0 10.802683 8.600000 -77387.465954 -77387.477585 -77387.466952 2.090801 2.318802 0.006467 -0.032436 1 0. 1 10.802683 8.600000 -77387.465340 -77387.492802 -77387.466489 2.093282 2.318802 0.006467 -0.032436 2 0. 2 10.802683 8.600000 -77387.464174 -77387.539134 -77387.465772 2.100792 2.318802 0.006467 -0.032436 3 0. 3 10.802683 8.600000 -77387.467889 -77387.582991 -77387.468670 2.108055 2.318802 0.006467 -0.032436 4 0. 4 10.802683 8.600000 -77387.467008 -77387.459389 -77387.466007 2.093235 2.318802 0.006467 -0.032436 5 0. 5 10.802683 8.600000 -77387.465826 -77387.432207 -77387.465538 2.099350 2.318802 0.006467 -0.032436 6 0. 6 10.802683 8.600000 -77387.465089 -77387.445521 -77387.465007 2.098870 2.318802 0.006467 -0.032436 7 0. 7 10.802683 8.600000 -77387.465332 -77387.460681 -77387.465192 2.098445 2.318802 0.006467 -0.032436 8 0. 8 10.802683 8.600000 -77387.465360 -77387.462474 -77387.465236 2.098569 2.318802 0.006467 -0.032436 9 0. 9 10.802683 8.600000 -77387.465362 -77387.461951 -77387.465233 2.098513 2.318802 0.006467 -0.032436 10 1. 0 10.802683 8.600000 -77387.460012 -77387.469787 -77387.460220 2.094443 2.095318 -0.045519 -0.033396 11 1. 1 10.802683 8.600000 -77387.460005 -77387.472545 -77387.460267 2.094928 2.095318 -0.045519 -0.033396 12 1. 2 10.802683 8.600000 -77387.459992 -77387.480820 -77387.460413 2.096383 2.095318 -0.045519 -0.033396 13 1. 3 10.802683 8.600000 -77387.459909 -77387.496109 -77387.460596 2.099164 2.095318 -0.045519 -0.033396 14 1. 4 10.802683 8.600000 -77387.459849 -77387.483230 -77387.460247 2.097002 2.095318 -0.045519 -0.033396 15 1. 5 10.802683 8.600000 -77387.459837 -77387.482892 -77387.460233 2.096926 2.095318 -0.045519 -0.033396 16 1. 6 10.802683 8.600000 -77387.459826 -77387.483043 -77387.460230 2.096961 2.095318 -0.045519 -0.033396 17 2. 0 10.802683 8.600000 -77387.459876 -77387.469584 -77387.459850 2.093987 2.095365 -0.047880 -0.020161 18 2. 1 10.802683 8.600000 -77387.459888 -77387.471608 -77387.459903 2.094331 2.095365 -0.047880 -0.020161 19 2. 2 10.802683 8.600000 -77387.459926 -77387.477680 -77387.460062 2.095364 2.095365 -0.047880 -0.020161 20 2. 3 10.802683 8.600000 -77387.459982 -77387.483555 -77387.460234 2.096339 2.095365 -0.047880 -0.020161 21 2. 4 10.802683 8.600000 -77387.460087 -77387.481466 -77387.460288 2.095811 2.095365 -0.047880 -0.020161 22 2. 5 10.802683 8.600000 -77387.460124 -77387.481317 -77387.460310 2.095700 2.095365 -0.047880 -0.020161 23 3. 0 10.802683 8.600000 -77387.460261 -77387.469926 -77387.460626 2.095151 2.095499 -0.048408 -0.024003 24 3. 1 10.802683 8.600000 -77387.460262 -77387.470303 -77387.460635 2.095216 2.095499 -0.048408 -0.024003 25 3. 2 10.802683 8.600000 -77387.460266 -77387.471434 -77387.460663 2.095410 2.095499 -0.048408 -0.024003 26 3. 3 10.802683 8.600000 -77387.460275 -77387.473579 -77387.460725 2.095763 2.095499 -0.048408 -0.024003 27 3. 4 10.802683 8.600000 -77387.460279 -77387.472274 -77387.460704 2.095511 2.095499 -0.048408 -0.024003 28 4. 0 10.802683 8.600000 -77387.460351 -77387.470022 -77387.460459 2.094666 2.095620 -0.045926 -0.027269 29 4. 1 10.802683 8.600000 -77387.460353 -77387.470562 -77387.460473 2.094760 2.095620 -0.045926 -0.027269 30 4. 2 10.802683 8.600000 -77387.460356 -77387.472183 -77387.460512 2.095043 2.095620 -0.045926 -0.027269 31 4. 3 10.802683 8.600000 -77387.460384 -77387.475853 -77387.460635 2.095674 2.095620 -0.045926 -0.027269 32 4. 4 10.802683 8.600000 -77387.460392 -77387.473435 -77387.460594 2.095216 2.095620 -0.045926 -0.027269 33 4. 5 10.802683 8.600000 -77387.460402 -77387.473051 -77387.460590 2.095111 2.095620 -0.045926 -0.027269 34 4. 6 10.802683 8.600000 -77387.460408 -77387.473298 -77387.460600 2.095147 2.095620 -0.045926 -0.027269 35 5. 0 10.802683 8.600000 -77387.460663 -77387.470346 -77387.460609 2.094289 2.095708 -0.049409 -0.033571 36 5. 1 10.802683 8.600000 -77387.460667 -77387.470932 -77387.460625 2.094389 2.095708 -0.049409 -0.033571 37 5. 2 10.802683 8.600000 -77387.460678 -77387.472692 -77387.460672 2.094688 2.095708 -0.049409 -0.033571 38 5. 3 10.802683 8.600000 -77387.460706 -77387.475868 -77387.460765 2.095216 2.095708 -0.049409 -0.033571 39 5. 4 10.802683 8.600000 -77387.460726 -77387.473877 -77387.460746 2.094822 2.095708 -0.049409 -0.033571 40 6. 0 10.802683 8.600000 -77387.460778 -77387.470485 -77387.461255 2.095445 2.095610 -0.048972 -0.036624 41 6. 1 10.802683 8.600000 -77387.460769 -77387.470152 -77387.461240 2.095391 2.095610 -0.048972 -0.036624 42 6. 2 10.802683 8.600000 -77387.460745 -77387.469155 -77387.461198 2.095231 2.095610 -0.048972 -0.036624 43 6. 3 10.802683 8.600000 -77387.460605 -77387.466208 -77387.461009 2.094845 2.095610 -0.048972 -0.036624 44 6. 4 10.802683 8.600000 -77387.460597 -77387.467710 -77387.461035 2.095142 2.095610 -0.048972 -0.036624 45 7. 0 10.802683 8.600000 -77387.460594 -77387.470275 -77387.461146 2.095728 2.095578 -0.039428 -0.040639 46 7. 1 10.802683 8.600000 -77387.460591 -77387.469916 -77387.461136 2.095666 2.095578 -0.039428 -0.040639 47 7. 2 10.802683 8.600000 -77387.460584 -77387.468840 -77387.461108 2.095482 2.095578 -0.039428 -0.040639 48 8. 0 10.802683 8.600000 -77387.460559 -77387.470247 -77387.461008 2.095405 2.095454 -0.039692 -0.039728 49 8. 1 10.802683 8.600000 -77387.460558 -77387.470382 -77387.461009 2.095429 2.095454 -0.039692 -0.039728 50 8. 2 10.802683 8.600000 -77387.460556 -77387.470785 -77387.461015 2.095503 2.095454 -0.039692 -0.039728 51 8. 3 10.802683 8.600000 -77387.460529 -77387.471589 -77387.461000 2.095678 2.095454 -0.039692 -0.039728 52 8. 4 10.802683 8.600000 -77387.460519 -77387.470835 -77387.460976 2.095560 2.095454 -0.039692 -0.039728 ...... 132 26. 6 10.802683 8.600000 -77387.460758 -77387.471728 -77387.461001 2.095030 2.095637 -0.036717 -0.048059 133 27. 0 10.802683 8.600000 -77387.460615 -77387.470313 -77387.461025 2.095406 2.095589 -0.038881 -0.050831 134 27. 1 10.802683 8.600000 -77387.460613 -77387.470035 -77387.461017 2.095359 2.095589 -0.038881 -0.050831 135 27. 2 10.802683 8.600000 -77387.460607 -77387.469200 -77387.460995 2.095217 2.095589 -0.038881 -0.050831
This log file shows 8 DFT+DMFT cycles, and at each cycle there are 4-10 DFT steps (column 3). The chemical potential (column 4) is fixed, as well as the double-counting (this is because we choose DCs='nominal' in params.dat file. We could choose DCs='exactd', which would give similar results, but would take more time to converge). The next two columns shows total energy and free energy. Finally columns 9 and 10 show the impurity and lattice occupancy, which should match once the loop is converged. The last two columns show the first and the last impurity level.
At this point, we can proceed to the next step.
Once the calculation is done on the imaginary axis, we need to do analytical continuation to obtain the self energy on the real axis. For this,we first take average of the sig.inp files from the last few steps (in order to reduce the noise) by
saverage.py sig.inp.30.?
maxent_run.py sig.inpx
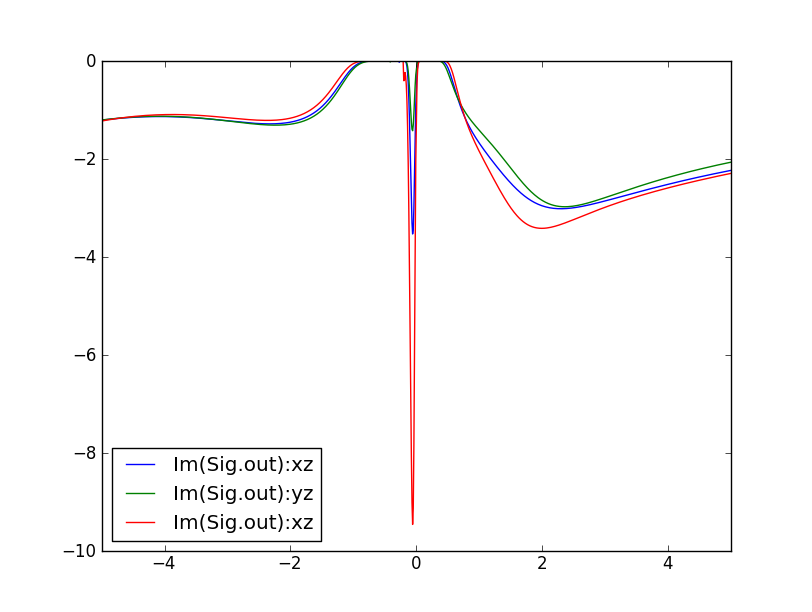
A few clarifications are in order. Such self-energy with a pole-like structure can not be analytically continued with maax-entropy method. The high-quality of this self-energy on real-axis is due to a trick that we use: We cobstruct a new auxiliary object, which has the following form Gt(i*w)=1/(i*w-Sigma(i*w)+Sigma(infty)). This quantity is analytic (as all other quantities like Sigma and G) and has a gap when self-energy diverges. Similarly to the DMFT green's function, it is quite well-behaved on the real axis, and can be easily continued to the real axis. Once we have Im(Gt(w)) on the real axis, we perform the Kramars-Kronig transformation to obtain complex Gt(w). From that, we get self-energy as Sigma(w)=w-1/Gt(w)+Sigma(infty).
Now we need to make one last dmft1 calculation in order to obtain the Green's function and DOS on the real axis. Create a new directory, and use "dmft_copy.py <dmft_results>" to copy necessary files from the output of the DMFT run to the new directory. Also copy the analytically continued self energy Sig.out to sig.inp to this new directory. Since we need to run the code on the real axis, we need to change the .indmfl file. The first number on the second line of .indmfl file determines whether the code is run on the real axis (0) or the imaginary axis (1). Change it to 0. Then, run
x lapw0 -f LaVO3 x_dmft.py lapw1 x_dmft.py dmft1
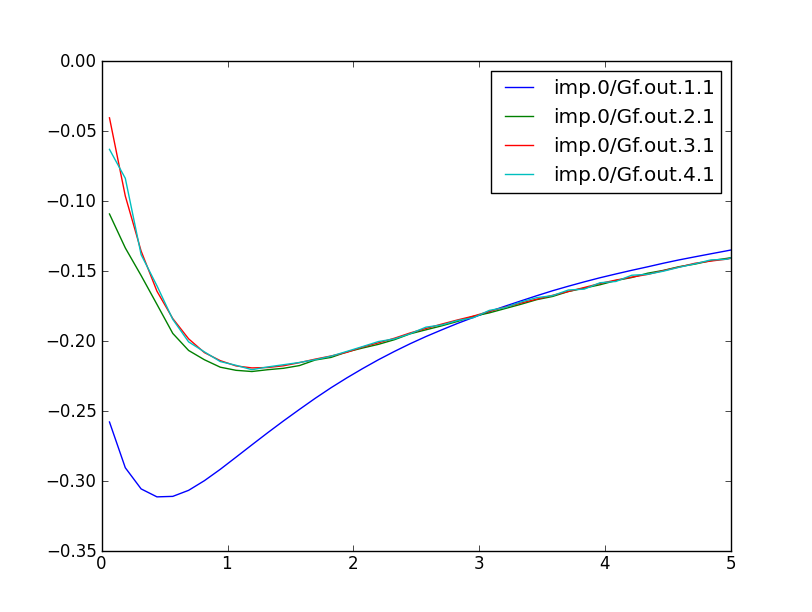
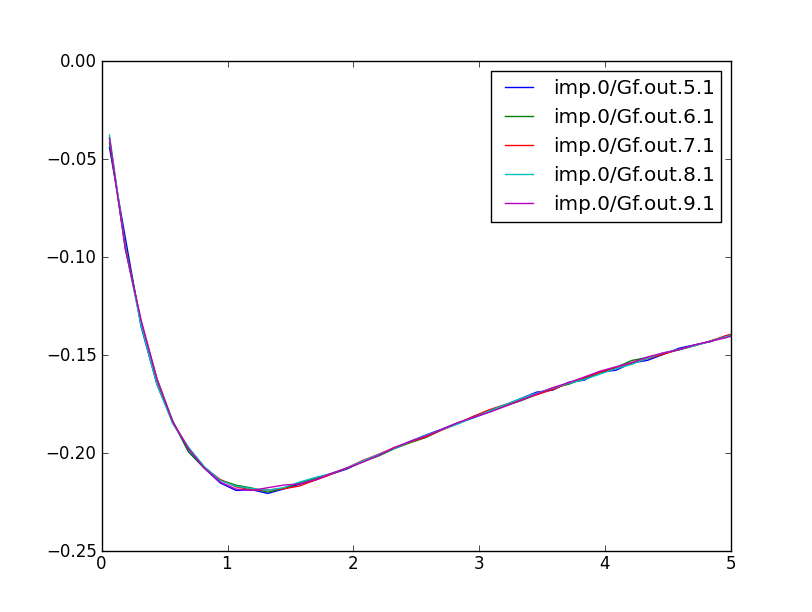
Once the run is done, we now have the densities of states on the real axis. Both the partial (V-d) and the total DOS is stored in the 'LaVO3.cdos' file:
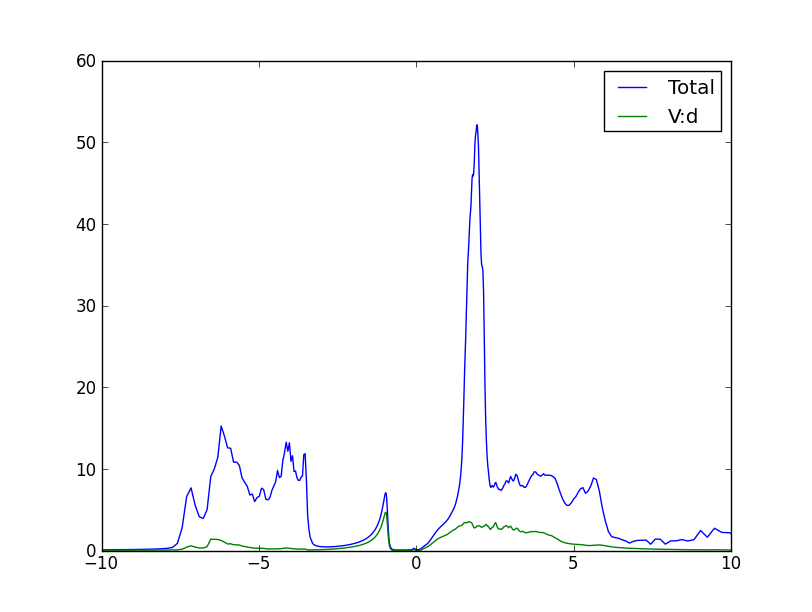
We can also take a look at the imaginary part of the green's function. Recall that the imaginary part of the green's function, divided by -pi, gives the DOS. So, plotting the even-numbered columns in the .gc1, we can get the DOS of individual d orbitals. It looks like this:
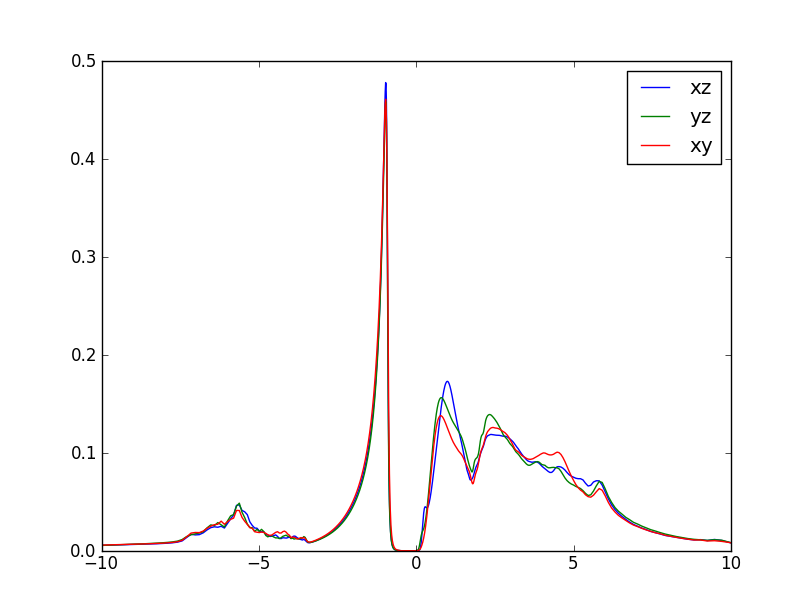
Another quantity of physical interest is the momentum resolved spectral function, which is an equivalent of spaghetti plot in DFT. First, we need to select a path in momentum space and store it into LaVO3.klist_band. One simple way is to open "xcrysden" and select option "File/Open-WIEN2k/Select-k-path". Alternatively, one could select from one of the Wien2k prepared templates. Next, we run Wien2k on the selected path, i.e.,
x lapw0 -f LaVO3 x_dmft.py lapw1 --band
0 0.025 0.025 200 -3.000000 3.000000 # matsubara, broadening-corr, broadening-noncorr, nomega, omega_min, omega_max (in eV)
x_dmft.py dmftp
echo 10.802683 > EF.dat
Next we execute
wakplot.py
wakplot.py 0.2
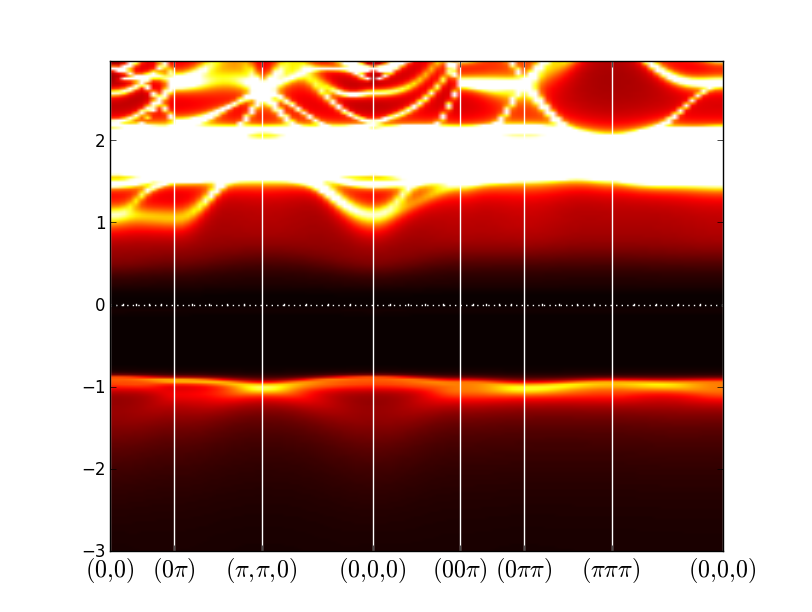
Note that the very bright set of bands around 2eV are La-f states.