
cif2struct.py SrVO3.cif
cif2struct.py -w -N 300 SrVO3.cif
Enter reduction in %: 0 Use old or new scheme (o/N) : N Do you want to accept these radii; .... (a/d/r) : a specify nn-bondlength factor: 2 Ctrl-X-C continue with sgroup : c Ctrl-X-C continue with symmetry : c Ctrl-X-C continue with lstart : c specify switches for instgen_lapw (or press ENTER): ENTER SELECT XCPOT: 5 SELECT ENERGY : -6 Ctrl-X-C continue with kgen : c Ctrl-X-C Ctrl-X-C NUMBER OF K-POINTS : 500 Shift of k-mesh (0=no, 1=shift) : 1 Ctrl-X-C c Ctrl-X-C n
run_lapw
x kgen
run_lapw
Specify correlated atoms (ex: 1-4,7,8): 1 Do you want to continue; or edit again? (c/e): c For each atom, specify correlated orbital(s) (ex: d,f): 1 V: d Do you want to continue; or edit again? (c/e): c Specify qsplit for each correlated orbital (default = 0): 1 V-1 d: 3 Do you want to continue; or edit again? (c/e): c Specify projector type (default = 2): 5 Do you want to continue; or edit again? (c/e): c Do you want to group any of these orbitals into cluster-DMFT problems? (y/n): n Enter the correlated problems forming each unique correlated problem, separated by spaces (ex: 1,3 2,4 5-8): 1 Do you want to continue; or edit again? (c/e): c Range (in eV) of hybridization taken into account in impurity problems; default -10.0, 10.0:Perform calculation on real; or imaginary axis? (r/i): i Ctrl-X-C Is this a spin-orbit run? (y/n): n Ctrl-X-C
There are 5 atoms in the unit cell: 1 V 2 Sr 3 O 4 O 5 O Specify correlated atoms (ex: 1-4,7,8): 1
For each atom, specify correlated orbital(s) (ex: d,f): 1 V: d
Specify qsplit for each correlated orbital (default = 0):
Qsplit Description
------ ------------------------------------------------------------
0 average GF, non-correlated
1 |j,mj> basis, no symmetry, except time reversal (-jz=jz)
-1 |j,mj> basis, no symmetry, not even time reversal (-jz=jz)
2 real harmonics basis, no symmetry, except spin (up=dn)
-2 real harmonics basis, no symmetry, not even spin (up=dn)
3 t2g orbitals
-3 eg orbitals
4 |j,mj>, only l-1/2 and l+1/2
5 axial symmetry in real harmonics
6 hexagonal symmetry in real harmonics
7 cubic symmetry in real harmonics
8 axial symmetry in real harmonics, up different than down
9 hexagonal symmetry in real harmonics, up different than down
10 cubic symmetry in real harmonics, up different then down
11 |j,mj> basis, non-zero off diagonal elements
12 real harmonics, non-zero off diagonal elements
13 J_eff=1/2 basis for 5d ions, non-magnetic with symmetry
14 J_eff=1/2 basis for 5d ions, no symmetry
------ ------------------------------------------------------------
1 V-1 d: 3
12 31 1 5 # hybridization band index nemin and nemax, renormalize for interstitials, projection type 1 0.025 0.025 200 -3.000000 1.000000 # matsubara, broadening-corr, broadening-noncorr, nomega, omega_min, omega_max (in eV) 1 # number of correlated atoms 1 1 0 # iatom, nL, locrot 2 3 1 # L, qsplit, cix
#================ # Siginds and crystal-field transformations for correlated orbitals ================ 1 5 3 # Number of independent kcix blocks, max dimension, max num-independent-components 1 5 3 # cix-num, dimension, num-independent-components #---------------- # Independent components are -------------- 'xz' 'yz' 'xy' #---------------- # Sigind follows -------------------------- 1 0 0 0 0 0 2 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 0 0 #---------------- # Transformation matrix follows ----------- 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 -0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 -0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000
The second line specifies the dimension of the first correlated block
(here "d" has dimension 5), and the three t2g components are treated
as independent.
Note that the orbitals, which have index "0" in the
Sigind block are treated statically. It is desirable to rearrange the
orbitals in such a way that the correlated orbitals are listed first,
followed by those, which are not correlated. This is already done
here, but if it was not, we would
permute the index table Sigind simultaneously with the rows
of Transformation matrix. Namely, each row in the
transformation matrix corresponds to a correlated orbital, for example row
4 is the z^2 orbital (equivalent to Y_{2,0}). As the forth diagonal
element in Sigind is set to zero, this orbital is not
correlated. The first three rows in Transformation matrix are
familiar xz,yz,xy orbitals, and are correlated in Sigind block.
Note also that the phase of each correlated orbital can be arbitrarily
chosen, and the physical observables should not depend on that choice. To keep compatibility
with Wien2k choice, we sometimes use i*xy, rather than xy orbital.
As explained above, we could alternatively use the symmetry and treat all
three t2g states as degenerate. In this case the indmfl would
take the form
#================ # Siginds and crystal-field transformations for correlated orbitals ================ 1 5 1 # Number of independent kcix blocks, max dimension, max num-independent-components 1 5 1 # cix-num, dimension, num-independent-components #---------------- # Independent components are -------------- 't2gs' #---------------- # Sigind follows -------------------------- 1 0 0 0 0 0 1 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 #---------------- # Transformation matrix follows ----------- ....
dmft_copy.py <lda_results_directory>
solver = 'CTQMC' # impurity solver
DCs = 'exact' # exact double-counting with dielectric constant approx.
max_dmft_iterations = 1 # number of iteration of the dmft-loop only
max_lda_iterations = 100 # number of iteration of the LDA-loop only
finish = 50 # number of iterations of full charge loop (1 = no charge self-consistency)
ntail = 300 # on imaginary axis, number of points in the tail of the logarithmic mesh
cc = 5e-5 # the charge density precision to stop the LDA+DMFT run
ec = 5e-5 # the energy precision to stop the LDA+DMFT run
recomputeEF = 1 # Recompute EF in dmft2 step. If recomputeEF = 0, it fixed the chemical potential. Good for insulators
#mix_delta = 0.5 # If instability occurs, we should start mixing hybridization
# Impurity problem number 0
iparams0={"exe" : ["ctqmc" , "# Name of the executable"],
"U" : [8.0 , "# Coulomb repulsion (F0)"],
"J" : [0.8 , "# Coulomb repulsion (F0)"],
"CoulombF" : ["'Ising'" , "# Form of Coulomb repulsion. 'Full' allows rotational invariant form of C.I."],
"beta" : [50 , "# Inverse temperature T=116K"],
"svd_lmax" : [25 , "# We will use SVD basis to expand G, with this cutoff"],
"M" : [5e6 , "# Total number of Monte Carlo steps"],
"mode" : ["SH" , "# We will use self-energy sampling, and Hubbard I tail"],
"nom" : [200 , "# Number of Matsubara frequency points sampled"],
"tsample" : [200 , "# How often to record measurements"],
"GlobalFlip" : [1000000 , "# How often to try a global flip"],
"warmup" : [3e5 , "# Warmup number of QMC steps"],
"nf0" : [1.0 , "# Nominal occupancy nd for double-counting"],
}
We also need a blank self-energy file (sig.inp), which we will generate by
szero.py
Now we can run the DFT+EDMFTF calculation. Do not forget to create files for mpi execution, which should be called mpi_prefix.dat and mpi_prefix.dat2. A command similar to the following, should create them:
echo "mpirun -np $NSLOTS " > mpi_prefix.dat $WIEN_DMFT_ROOT/createDMFprefix.py $JOBNAME.klist mpi_prefix.dat > mpi_prefix.dat2
$WIEN_DMFT_ROOT/run_dmft.py > nohup.dat 2>&1
export WIENROOT=<wien-instalation-dir> export WIEN_DMFT_ROOT=<dir-containing-dmft-executables> export SCRATCH="." export PYTHONPATH=$PYTHONPATH:$WIEN_DMFT_ROOT
export PATH=$PATH:$WIENROOT:$WIEN_DMFT_ROOT
Sat Mar 25 20:27:19 EDT 2017> (x) -f SrVO3 lapw0 Sat Mar 25 20:27:37 EDT 2017> lapw1 Sat Mar 25 20:27:39 EDT 2017> dmft1 Sat Mar 25 20:27:42 EDT 2017> impurity Sat Mar 25 20:30:45 EDT 2017> dmft2 Sat Mar 25 20:30:48 EDT 2017> (x) -f SrVO3 lcore Sat Mar 25 20:30:49 EDT 2017> (x) -f SrVO3 mixer Sat Mar 25 20:30:50 EDT 2017> (x) -f SrVO3 lapw0 Sat Mar 25 20:30:53 EDT 2017> lapw1 Sat Mar 25 20:30:55 EDT 2017> dmft2 Sat Mar 25 20:30:58 EDT 2017> (x) -f SrVO3 lcore Sat Mar 25 20:30:59 EDT 2017> (x) -f SrVO3 mixer Sat Mar 25 20:31:00 EDT 2017> (x) -f SrVO3 lapw0 Sat Mar 25 20:31:03 EDT 2017> lapw1 Sat Mar 25 20:31:05 EDT 2017> dmft2 Sat Mar 25 20:31:08 EDT 2017> (x) -f SrVO3 lcore Sat Mar 25 20:31:09 EDT 2017> (x) -f SrVO3 mixer Sat Mar 25 20:31:10 EDT 2017> (x) -f SrVO3 lapw0 Sat Mar 25 20:31:12 EDT 2017> lapw1 Sat Mar 25 20:31:15 EDT 2017> dmft2 Sat Mar 25 20:31:19 EDT 2017> (x) -f SrVO3 lcore Sat Mar 25 20:31:20 EDT 2017> (x) -f SrVO3 mixer Sat Mar 25 20:31:21 EDT 2017> (x) -f SrVO3 lapw0 Sat Mar 25 20:31:23 EDT 2017> lapw1 Sat Mar 25 20:31:25 EDT 2017> dmft2 Sat Mar 25 20:31:29 EDT 2017> (x) -f SrVO3 lcore Sat Mar 25 20:31:30 EDT 2017> (x) -f SrVO3 mixer Sat Mar 25 20:31:31 EDT 2017> (x) -f SrVO3 lapw0 Sat Mar 25 20:31:33 EDT 2017> lapw1 Sat Mar 25 20:31:35 EDT 2017> dmft2 Sat Mar 25 20:31:38 EDT 2017> (x) -f SrVO3 lcore Sat Mar 25 20:31:39 EDT 2017> (x) -f SrVO3 mixer Sat Mar 25 20:31:40 EDT 2017> (x) -f SrVO3 lapw0 Sat Mar 25 20:31:43 EDT 2017> lapw1 Sat Mar 25 20:31:45 EDT 2017> dmft2 Sat Mar 25 20:31:49 EDT 2017> (x) -f SrVO3 lcore Sat Mar 25 20:31:49 EDT 2017> (x) -f SrVO3 mixer Sat Mar 25 20:31:51 EDT 2017> (x) -f SrVO3 lapw0 ..... Sat Mar 25 20:32:50 EDT 2017> (x) -f SrVO3 lapw0 Sat Mar 25 20:32:53 EDT 2017> lapw1 Sat Mar 25 20:32:55 EDT 2017> dmft1 Sat Mar 25 20:32:57 EDT 2017> impurity Sat Mar 25 20:34:39 EDT 2017> dmft2 Sat Mar 25 20:34:43 EDT 2017> (x) -f SrVO3 lcore Sat Mar 25 20:34:43 EDT 2017> (x) -f SrVO3 mixer Sat Mar 25 20:34:45 EDT 2017> (x) -f SrVO3 lapw0
:CHARGE convergence: 1.0 :CHARGE convergence: 0.0835935 :CHARGE convergence: 0.0439781 :CHARGE convergence: 0.0657028 :CHARGE convergence: 0.0459584 :CHARGE convergence: 0.0432044 :CHARGE convergence: 0.0043898 :CHARGE convergence: 0.0063747 :CHARGE convergence: 0.0034868 :CHARGE convergence: 0.0016732 :CHARGE convergence: 0.0003244 :CHARGE convergence: 5.52e-05 :CHARGE convergence: 1.36e-05 :CHARGE convergence: 0.0052109 :CHARGE convergence: 0.0045152 :CHARGE convergence: 0.0025189 :CHARGE convergence: 0.0027001 :CHARGE convergence: 0.0007694 :CHARGE convergence: 0.0004947 :CHARGE convergence: 0.0001302 :CHARGE convergence: 3.72e-05
:CHARGE convergence: 6.4e-05 :CHARGE convergence: 5.16e-05 :CHARGE convergence: 7.82e-05 :CHARGE convergence: 3.24e-05 :CHARGE convergence: 2.87e-05 :CHARGE convergence: 3.05e-05 :CHARGE convergence: 2.62e-05 :CHARGE convergence: 4.31e-05 :CHARGE convergence: 3.77e-05 :CHARGE convergence: 5.45e-05 :CHARGE convergence: 4.65e-05 :CHARGE convergence: 5.75e-05 :CHARGE convergence: 4.95e-05 :CHARGE convergence: 5.02e-05 :CHARGE convergence: 4.25e-05 :CHARGE convergence: 9.4e-06 :CHARGE convergence: 8.2e-06
# #. # mu Vdc Etot Ftot+T*Simp Ftot+T*Simp n_latt n_imp Eimp[0] Eimp[-1] 0 0. 0 8.770334 3.881170 -8696.600092 -8696.627108 -8696.589740 1.390867 1.035714 -0.042696 -0.042696 1 0. 1 8.721143 3.881170 -8696.599194 -8696.671599 -8696.589452 1.403287 1.035714 -0.042696 -0.042696 2 0. 2 8.573921 3.881170 -8696.598217 -8696.803391 -8696.590010 1.441857 1.035714 -0.042696 -0.042696 3 0. 3 8.202136 3.881170 -8696.605608 -8697.122541 -8696.601390 1.551790 1.035714 -0.042696 -0.042696 4 0. 4 8.460228 3.881170 -8696.598488 -8696.841630 -8696.590804 1.460942 1.035714 -0.042696 -0.042696 5 0. 5 8.147676 3.881170 -8696.605366 -8697.102050 -8696.601312 1.556594 1.035714 -0.042696 -0.042696 6 0. 6 8.373934 3.881170 -8696.600005 -8696.929838 -8696.593972 1.498405 1.035714 -0.042696 -0.042696 7 0. 7 8.317077 3.881170 -8696.600855 -8696.972107 -8696.595356 1.511839 1.035714 -0.042696 -0.042696 8 0. 8 8.325935 3.881170 -8696.600578 -8696.965302 -8696.594961 1.508504 1.035714 -0.042696 -0.042696 9 0. 9 8.333896 3.881170 -8696.600401 -8696.958379 -8696.594737 1.506428 1.035714 -0.042696 -0.042696 10 0. 10 8.339930 3.881170 -8696.600365 -8696.953237 -8696.594558 1.504693 1.035714 -0.042696 -0.042696 11 0. 11 8.341431 3.881170 -8696.600332 -8696.952012 -8696.594515 1.504284 1.035714 -0.042696 -0.042696 12 0. 12 8.341274 3.881170 -8696.600336 -8696.952123 -8696.594520 1.504328 1.035714 -0.042696 -0.042696 13 1. 0 8.309814 4.371950 -8696.593956 -8696.597300 -8696.590861 1.480979 1.114940 -0.789484 -0.789484 14 1. 1 8.307926 4.371950 -8696.594006 -8696.598954 -8696.590931 1.481605 1.114940 -0.789484 -0.789484 15 1. 2 8.302265 4.371950 -8696.594162 -8696.603913 -8696.591144 1.483484 1.114940 -0.789484 -0.789484 16 1. 3 8.289969 4.371950 -8696.594473 -8696.614043 -8696.591648 1.487772 1.114940 -0.789484 -0.789484 17 1. 4 8.298014 4.371950 -8696.594307 -8696.601976 -8696.591415 1.485486 1.114940 -0.789484 -0.789484 18 1. 5 8.295568 4.371950 -8696.594356 -8696.604601 -8696.591467 1.486003 1.114940 -0.789484 -0.789484 19 1. 6 8.294026 4.371950 -8696.594367 -8696.605882 -8696.591468 1.486078 1.114940 -0.789484 -0.789484 20 1. 7 8.294637 4.371950 -8696.594359 -8696.605625 -8696.591463 1.486020 1.114940 -0.789484 -0.789484 21 2. 0 8.320967 4.713672 -8696.589028 -8696.596377 -8696.586800 1.475189 1.169843 -0.763354 -0.763353 ...... 195 29. 0 8.480065 6.490811 -8696.565894 -8696.573610 -8696.565344 1.449904 1.445104 -1.067816 -1.067892 196 29. 1 8.480403 6.490811 -8696.565884 -8696.573243 -8696.565331 1.449812 1.445104 -1.067816 -1.067892 197 29. 2 8.481417 6.490811 -8696.565852 -8696.572142 -8696.565292 1.449536 1.445104 -1.067816 -1.067892 198 29. 3 8.487627 6.490811 -8696.565668 -8696.564739 -8696.565066 1.447849 1.445104 -1.067816 -1.067892 199 29. 4 8.482462 6.490811 -8696.565826 -8696.569727 -8696.565259 1.449233 1.445104 -1.067816 -1.067892 200 29. 5 8.482355 6.490811 -8696.565827 -8696.569881 -8696.565260 1.449244 1.445104 -1.067816 -1.067892 201 29. 6 8.482271 6.490811 -8696.565825 -8696.570101 -8696.565258 1.449240 1.445104 -1.067816 -1.067892 202 30. 0 8.487685 6.495397 -8696.568237 -8696.576298 -8696.566507 1.446473 1.446400 -1.065371 -1.065310 203 30. 1 8.487229 6.495397 -8696.568251 -8696.576779 -8696.566524 1.446595 1.446400 -1.065371 -1.065310 204 30. 2 8.485860 6.495397 -8696.568292 -8696.578220 -8696.566575 1.446962 1.446400 -1.065371 -1.065310 205 30. 3 8.477397 6.495397 -8696.568550 -8696.587772 -8696.566884 1.449201 1.446400 -1.065371 -1.065310 206 30. 4 8.484230 6.495397 -8696.568333 -8696.581241 -8696.566624 1.447393 1.446400 -1.065371 -1.065310 207 30. 5 8.484380 6.495397 -8696.568332 -8696.581067 -8696.566624 1.447383 1.446400 -1.065371 -1.065310 208 30. 6 8.484556 6.495397 -8696.568334 -8696.580786 -8696.566626 1.447377 1.446400 -1.065371 -1.065310 209 31. 0 8.483446 6.518435 -8696.565292 -8696.572554 -8696.564673 1.449912 1.448534 -1.072144 -1.072250
The comparison on nlatt (column 9) and nimp (column 10) is a very non-trivial check of numerics precision and convergence, as the equivalence between the local and the impurity Green's function is not imposed, but is achieved only once the self-consistency is reached. If you'd like to get more information about the details of the run, you can also monitor the files SrVO3.scf, SrVO3.outputdmf1, SrVO3.outputdmf2, dmft_info, dmft1_info.out, dmft2_info.out, imp.0/ctqmc.log, imp.0/nohup_imp.out.xxx, and many others.
Notice that most of the parameters in "params.dat" file can be changed
during the run, and their value will be updated in the next start of
the global loop. For
example, one can change the number of charge steps
("max_lda_iterations") or dmft steps ("max_dmft_iterations") or number
of Monte Carlo steps ("M") during the run to get convergence
faster. It does take some experience to choose efficient set of parameters.
Sometimes a double-period appears, and the self-energy is jumping between two different values from iteration to iteration. Or the self-energy starts to jump from iteration to iteration once it was almost converged. In this case, we should mix hybridization function and the impurity levels. This is achieved by specifying
mix_delta = 0.5
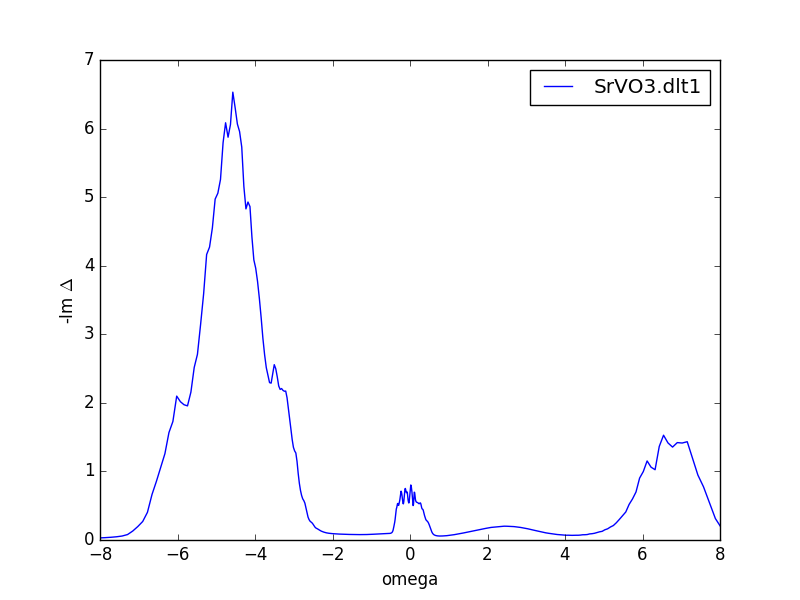
As mentioned above, the fastest way to check convergence is to plot the impurity and the lattice occupancy in column 9 and 10 of info.iterate
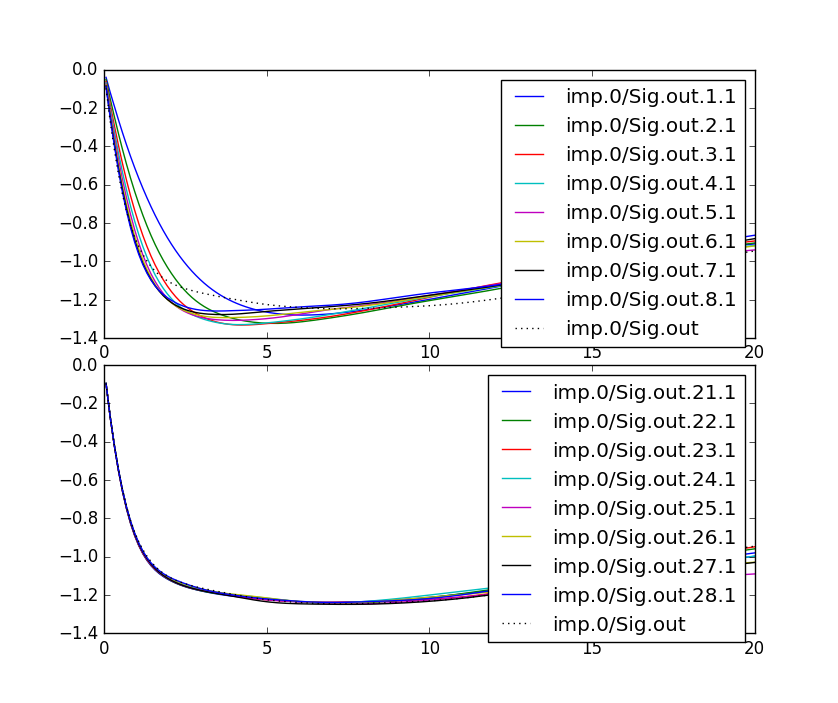
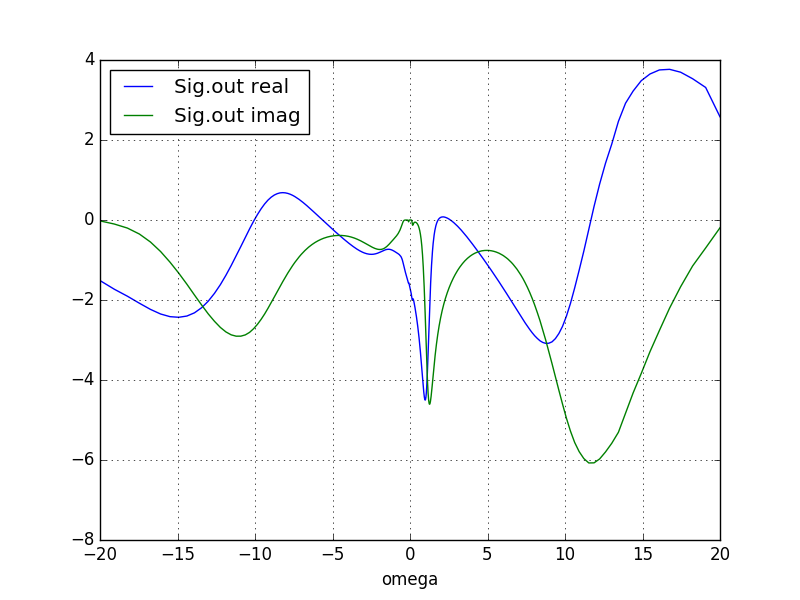
Another way to check the convergence is to plot the self-energy.
The impurity self energy is stored in the 'imp.0/Sig.out.*.*' file in
the. In our case, this file has seven columns: The first
column is the energy, and there is a pair of columns (real and
imaginary) for each orbital. Plotting the imaginary part of first few
iterations, we get:
saverage.py sig.inp.5?
params={'statistics': 'fermi', # fermi/bose
'Ntau' : 400, # Number of time points
'L' : 20.0, # cutoff frequency on real axis
'x0' : 0.01, # low energy cut-off
'bwdth' : 0.004, # smoothing width
'Nw' : 300, # number of frequency points on real axis
'gwidth' : 2*15.0, # width of gaussian
'idg' : 1, # error scheme: idg=1 -> sigma=deltag ; idg=0 -> sigma=deltag*G(tau)
'deltag' : 0.001, # error
'Asteps' : 4000, # anealing steps
'alpha0' : 1000, # starting alpha
'min_ratio' : 0.001, # condition to finish, what should be the ratio
'iflat' : 1, # iflat=0 : constant model, iflat=1 : gaussian of width gwidth, iflat=2 : input using file model.dat
'Nitt' : 1000, # maximum number of outside iterations
'Nr' : 0, # number of smoothing runs
'Nf' : 40, # to perform inverse Fourier, high frequency limit is computed from the last Nf points
}
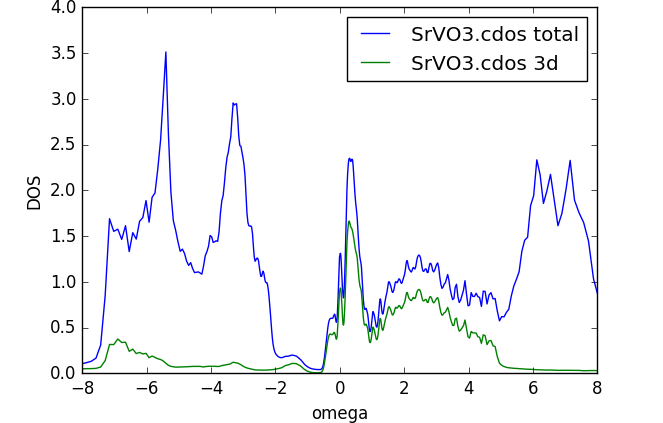
maxent_run.py sig.inpx
dmft_copy.py <dmft_results>
x lapw0 -f SrVO3 x_dmft.py lapw1 x_dmft.py dmft1
x kgen -f SrVO3
12 31 1 5 # hybridization band index nemin and nemax, renormalize for interstitials, projection type 0 0.025 0.05 200 -3.000000 1.000000 # matsubara, broadening-corr, broadening-noncorr, nomega, omega_min, omega_max (in eV) 1 # number of correlated atoms 1 1 0 # iatom, nL, locrot 2 3 1 # L, qsplit, cix #================ # Siginds and crystal-field transformations for correlated orbitals ================ 1 5 4 # Number of independent kcix blocks, max dimension, max num-independent-components 1 5 4 # cix-num, dimension, num-independent-components #---------------- # Independent components are -------------- 'xz' 'yz' 'xy' 'eg' #---------------- # Sigind follows -------------------------- 1 0 0 0 0 0 2 0 0 0 0 0 3 0 0 0 0 0 4 0 0 0 0 0 4 #---------------- # Transformation matrix follows ----------- 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 -0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 -0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000
# s_oo= [9.3552102519215712, 9.3552102519215712, 9.3552102519215712, 0] # Edc= [6.8617042077680876, 6.8617042077680876, 6.8617042077680876, 0]
x_dmft.py dmft1
12 31 1 4 # hybridization band index nemin and nemax, renormalize for interstitials, projection type 0 0.025 0.025 200 -3.000000 1.000000 # matsubara, broadening-corr, broadening-noncorr, nomega, omega_min, omega_max (in eV) 2 # number of correlated atoms 1 1 0 # iatom, nL, locrot 2 3 1 # L, qsplit, cix 3 1 0 # iatom, nL, locrot for oxygen states 1 1 0 # L, qsplit, cix for oxygen states #================ # Siginds and crystal-field transformations for correlated orbitals ================ 1 5 3 # Number of independent kcix blocks, max dimension, max num-independent-components 1 5 3 # cix-num, dimension, num-independent-components #---------------- # Independent components are -------------- 'xz' 'yz' 'xy' #---------------- # Sigind follows -------------------------- 1 0 0 0 0 0 2 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 0 0 #---------------- # Transformation matrix follows ----------- 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 -0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 -0.00000000 -0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.70710679 0.00000000